




Rapid Marker Assessment of two Medicinal Plants and Thier Herbal Products used in the Management of Diabetes in Nasarawa State and the FCT
Mowobi Gbolaha Gabriel* and Onovo Josiah Chukwudi
Department of Plant Science and Biotechnology, Nasarawa State University, Nigeria
Submission: February 15, 2018; Published: April 26, 2018
*Corresponding author: Mowobi Gbolaha Gabriel, Plant Science and Biotechnology, Faculty of Natural and Applied Sciences, Nasarawa State University, PMB, 1022, Keffi Nigeria, Tel: +234 8056155336; Email: mowobig@gmail.com
How to cite this article: Mowobi G G, Onovo J C. Rapid Marker Assessment of two Medicinal Plants and Thier Herbal Products used in the Management of Diabetes in Nasarawa State and the FCT. Nutri Food Sci Int J. 2018; 6(3): 555689. DOI: 10.19080/NFSIJ.2018.06.555689
Abstract
Khaya senegalensis and Azadirachta indica are common plants that have been used in the management of diabetes. In the present investigation, RAPD Marker Assessment was carried out on Herbal Products from Khaya senegalensis and Azadirachta indica being used in the Management of Diabetes in Nasarawa State and Federal Capital Territory (FCT). Eleven decamer oligonucleotide primers were screened in the RAPD analysis for authentication of claim for genuine and adulterant samples. The Deoxyribonuclic Acid (DNA) isolated from the dried herbal samples was used as templates in polymerase chain reactions with the eleven primers. Five of the eleven primers gave species specific reproducible unique amplicons. The unique amplicons obtained in PCR amplification and the data generated from the amplification of the DNA were subjected to Numerical Taxonomy System of Statistic (NTSYS). From the phylogenetic tree generated among the 24 samples based on PCR product, it was observed that the relatedness was 37.5%. The analysis also showed that there was no strong interrelationship between herbal samples and plant sample at that index. At the end of the investigation, the result showed that the herbs contained both Khaya senegalensis and Azardirachta indica as they claim though; the quantity is low (37.5%).
Keywords: Khaya senegalensis; Azadirachta indica; Diabetes; Herbal product; RAPD markers; Polymorphic; Polyherbal; Taxonomy
Abbreviations: RAPD: Random Amplified Polymorphic DNA; DNA : Deoxyribonuclic Acid; NTSYS: Numerical Taxonomy System of Statistic; PHF: Polyherbal formulations; SDS: Sodium Dodecyl Sulfate; NaCl: Sodium Chloride; EDTA: Ethylenediaminetetraacetic acid; PVP: Polyvinylpyrolidione; CIA: Chloroform Isoamylalchohol; TE: Tris base EDTA; TBE: Tris base, boric acid and EDTA; UV: Ultraviolet; PCR: Polymerase Chain Reaction; UPGMA: Unweighted Pair Clustering Group Arithmetic Average; SHAN: Sequential Hierarchical Numeric.
Introduction
Many plants have been used for herbal and medicinal purposes not only by humans since prehistoric times [1,2], but are also used to treat various ailments by our closest relatives, the African great apes [3,4]. Up on till now, medicinal plants constitute the source of a large number of chemical compounds used as drugs in Western medicine and serve as the primary therapeutic resource for most of the world's population living in developing countries. The use of herbal preparations for health care purposes is also gaining popularity in developed countries at the same time [5]. The increased demand for botanical products is met by an expanding industry and accompanied by calls for assurance of quality, efficacy and safety [6]. Many ethnobotanical surveys on medicinal plants used by the local population have been performed in different parts of the world including Nigeria, China, Morroco, Saudi Arabia, Taiwan, and Trinidad and Tobago etc [7-9].
Herbal medicines are often used as therapeutic remedies in combination with allopathic drugs. Most of the doctors did not report any complication, but nausea, vomiting, gastric problems were the common adverse effects reported with Polyherbal formulations (PHF). Usually ayurvedic drugs are being used due to their minimum toxicity. Practicing physicians expected that controlled clinical trials of the herbal antidiabetics should be conducted in humans at different hospitals to substantiate the efficacy claim. Herbal medicines can be relevant today only if they are applied and tested within the framework of modern sciences and subjected to the rigorous criteria for quality, safety and efficacy. Only then, herbal products can be comparable with modern medicines and can bring necessary confidence in prescribing doctors. Major hindrance in amalgamation of herbal medicine in modern medical practices is lack of scientific and clinical data proving their efficacy and safety. There is a need for conducting clinical research in herbal drugs, developing simple bioassays for biological standardization, pharmacological and toxicological evaluation, and developing various animal models for toxicity and safety evaluation. It is also important to establish the active components from these plant extracts [10].
Diabetes mellitus is a complex metabolic disorder resulting from either insulin insufficiency or insulin dysfunction. Type I diabetes (insulin dependent) is caused due to insulin insufficiency because of lack of functional beta cells. Patients suffering from this are therefore totally dependent on exogenous source of insulin while patients suffering from Type II diabetes (insulin independent) are unable to respond to insulin and can be treated with dietary changes, exercise and medication [11]. Type II diabetes is the more common form of diabetes constituting 90% of the diabetic population [11].
Medicinal plants are being looked up once again for the treatment of diabetes. Many conventional drugs have been derived from prototypic molecules in medicinal plants. Metformin exemplifies an efficacious oral glucose-lowering agent. Its development was based on the use of Galega officinalis to treat diabetes. Galega officinalis is rich in guanidine, the hypoglycemic component. Because guanidine is too toxic for clinical use, the alkyl biguanides synthalin A and synthalin B were introduced as oral anti-diabetic agents in Europe in the 1920s but were discontinued after insulin became more widely available [10]. However, experience with guanidine and biguanides prompted the development of metformin. To date, over 400 traditional plant treatments for diabetes have been reported, although only a small number of these have received scientific and medical evaluation to assess their efficacy. The hypoglycemic effect of some herbal extracts has been confirmed in human and animal models of type II diabetes [10]. The World Health Organization Expert Committee on diabetes has recommended that traditional medicinal herbs be further investigated.
Khaya senegalensis belongs to the Family Meliaceae and is also known in French as Cailcedrat and African mahogany in English. It is a tree with shining foliage and wide dense crown; tall with height frequently between 15 to 20m but grows up to 35m. Mean average height was found to be 15m with an average of 54cm diameter and the trunk can be without branches for up to 10m. The bark is greyish and scaly and new leaves emerge during the dry season (December). Fruits is 4-6cm in diameter, Leaflets usually appear in drying pale glaucous green, Sepals is pale green while petals and staminal tube is cream and stigma appears yellow [12]. Khaya senegalensis occur from Mauritania and Senegal east to northern Uganda and it is commonly planted in its natural area of distribution, mainly as ornamental and roadside tree, and also outside this area e.g. Tanzania, Egypt, India, South Africa, Cape Verde, Malawi, Madagascar, Indonesia, Vietnam, Australia and Tropical America [12].
Azadirachta indica also known in English as neem and margosa belongs to the family Meliaceae [13]. It is indigenous to the dry forests of South and Southeast Asia and is widely distributed in India, Nepal, Pakistan, Bangladesh, Sri Lanka, Myanmar, Thailand, Malaysia, Indonesia and Iran. Original natural distribution is obscured by widespread cultivation but neem is believed to be native to at least Burma and NE India. The tree is adapted to hot and dry climates and that has made it one of the most commonly planted species in arid and semi-arid areas within its natural range and in Central and Southern America, the Caribbean, Philippines and Africa [13]. Azadirachta indica is a fast-growing tree that can reach a height of 15-20m; occasionally it reaches 35-40m. It has compound leaves imparipinnate, shinning, and deeply serrate; leaflets are sub-opposite, very oblique at base evergreen, but in severe drought it may shed most or nearly all of its leaves. It has a cream flower or yellowish white auxiliary, panicles is elongated. It fruits is one-seeded drupes 1-2cm long with woody endocarp, greenish yellow when ripe. Seeds is ellipsoid, cotyledons is thick, fleshy and oily [14].
Molecular approach has proved itself an increasingly valuable tool in the identification of plant varieties [15]. Some of the commercially desirable plant is reproduced by micro propagation; therefore all individuals belonging to a given variety share an identical genome. Lack of intravariety genetic variability greatly simplifies the task of identification because any difference between two given individuals unambiguously indicates that they belong to different varieties. One of the most successful techniques in this field is random amplified polymorphic DNA (RAPD) [16,17]. It has two main advantages: it allows random sampling of markers over whole genomic DNA and does not require any previous information on the genome of the organism under investigation.
PCR-based markers involve amplification of particular DNA loci, with the help of specific or arbitrary oligonucleotide primers and a thermostable DNA polymerase enzyme [18]. The major advantages of PCR techniques are that mainly only a small amount of DNA is required, no prior sequence information is needed, and many genetic markers can be generated within a short time.
The specific objective of this research is to use amplifiable RAPD markers from khaya senegalensis and Azadrachta indica to characterize the herbal products from Nasarawa State and F.C.T Nigeria.
Material and Methods
Study Area: The research work covers Nasarawa State and the Federal Capital Territory (FCT) Abuja Nigeria both of which are located in the middle belt of Nigeria. Nasarawa State is geographically situated on latitude of 8°5' N and longitude 7°5' E. The temperature range is between 25 °C - 31 °C and about 602mm of precipitation falls annually [19]. Abuja is located in the Guinea savannah. Raining season begins from April and ends in October in Abuja and the average day time temperature reach 28 °C to 30 °C. In dry season, average daily temperature can soar as high as 40 °C while Annual rainfall ranges from 1100mm to 1600mm. Gwagwalada is geographically located on latitude 8°56’59”N of the Equator and longitude 7°5’59” E [20]. The lab work was conducted at Biotechnology Advance Research Center, Sheda Science and Technology Complex (SHESTCO) Abuja, Nigeria.
Collection and storage of herbs/plants
A total of 12 samples (from Nasarawa State and from Federal Capital Territory) each of both plants (Khaya senegalensis and Azadirachta indica) and herbs made from them were assessed. Young leaves of both plants were harvested, appropriately labeled and Stored in the freezer at -20 °C. The leaves were used for DNA extraction, while excess leaf materials were stored for future DNA extraction.
Six (6) of the samples were collected from the FCT (Zuba, Giri, Kubwa, Orozo, Nyanyan and Apo Village) and six (6) from Nasarawa State (Garaku, Keffi, Uke, Kaduna Road, Auta and Nasarawa) to allow for a good representation of Nasarawa State and the FCT. Both plant and herbal sample where collected in the same location. Collected herbal samples were stored in a cool and dry place at room temperature (28 ± 2 °C) and also for further DNA extraction.
Genomic DNA extraction
Modified SDS method: Collected leaves were crushed in liquid nitrogen with mortar and pestle and 0.2g of both samples was weighed into 2ml eppendorf tube, 700μl of the extraction buffer (CTAB 2%, 1.4M NaCl, 100 mMTrisph 8.0, 20mM EDTA PH 8.0, 2% PVP, 20ul β-mercaptoethanol) pH 8.0 was added, it was incubated at 65 °C in heating block for 2 hours; it was cooled for 5-10 minutes at room temperature, Chloroform Isoamylalchohol (24:1) 600μl was added in the fume hood, it was mixed gently by inversion for 5 minutes. It was centrifuged for 5 minutes at 14000rpm. The supernatant was then transferred into fresh tubes, 500μl of ice cold isopropanol was added and mixed well gently by inversion for DNA precipitation. It was then incubated at -20 °C for 30 overnight; it was centrifuged at 2,000rpm for 15minutes at 4 °C. The supernatant was decanted in the fume hood, the residue was washed with 100μl 70% ethanol, procedure 12 above was repeated, the pellet was dried at room temperature. The dry pellet was redesolve in 50TE buffer and 2μl of RNase was added and stored at -4 °C [21].
Modified CTAB method: In 2x CTAB buffer (CTAB 2%, 1.4M NaCl, 100mM Trisph 8.0, 20mM EDTA PH 8.0, 1% PVP, 0.1% β-mercaptoethanol), about 1.3μl Mercaptoethanol and a pinch of PVP was added (CTAB buffer was pre heated at 65 °C), 1g of herbal powder was weighed into 2 ml extraction tubes. 700μl of CTAB buffer was added in number 2 above and incubated at 65 °C for 1 hour. Between times, vigorous vortexing was done for a good mix. 500μl of chloroform/ isoamylalchohol (24:1) was added and mix for 5mins, sample was Centrifuge at 14,000rpm for 5mins. Liquid phase was removed into a new 1.5ml tube, 500μl of ice cold isopropanol was added then it was incubated at - 20 °C for about 30min to 1 hour. It was then centrifuge for 2min at 14,000rpm, liquid was removed carefully so as not to throw away DNA rings at the bottom of the tube. 100μl of 70% ethanol was added to wash DNA, and then it was spin for 2min at 14,000rpm, ethanol was removed and number 10 above was repeated. DNA pellet was dry at room temperature for the ethanol to evaporate, 100μl of 1x TE buffer and 2μl RNase was added [22].
Modified CTAB method 2: In 2x CTAB buffer (CTAB 2%, 1.4M NaCl, 100mM Trisph 8.0, 20mM EDTA PH 8.0, 1% PVP, 0.1% β-mercaptoethanol), about 1.3μl Mercaptoethanol and a pinch of PVP was added (CTAB buffer was pre heated at 65 °C). 1g of herbal powder was weighed into 2 ml extraction tubes, 700μl of CTAB buffer was added in number 2 above and incubated at 65 °C for 1 hour. Between times, vigorous vortexing will be done for a good mix. 500μl of chloroform/ isoamylalchohol (24:1) was added and mix for 5mins, sample was Centrifuge at 14,000rpm for 5min, and liquid phase was remove into a new 1.5ml tube. 500of ice cold isopropanol was added then it was incubated at - 20 °C for about 30min to 1 hour. It was then centrifuge for 2min at 14,000rpm. The liquid was removed carefully so as not to throw away DNA rings at the bottom of the tube. 100μl of 70% ethanol was added to wash DNA, then it was spin for 2mins at 14,000 rpm. Ethanol was removed and number 10 above was repeated, DNA pellet was dry at room temperature for the ethanol to evaporate. 10 of 5M NaCl was added to the dry DNA pellet, 73μl of absolute (100%) ethanol was also added and then votexed, the sample was incubated on ice for 10 minute. It was then centrifuge at 10,000rpm for 15 minute, the solution was then removed in a new tube. 300μl of Isopropanol was added for precipitation. It was then incubated at - 20 °C for 1 hour. It was centrifuge at 10,000Rpm for 15 minute to remove the supernatant. It was then washed with 70% ethanol, DNA pellet was dry at room temperature for the ethanol to evaporate. 50of 1x TE buffer and 2μl, RNase was added [22].
Estimation of quality and quantity of isolated genomic DNA
Agarose gel electrophoresis: Fifty millilitres (50ml) of 1% (w/v) of agarose gel were prepared by weighing accurately 0.5g, of agarose powder (BDH Biochemical England) and made up to 50mls with X1 TBE (Trisma base- Boric acid- EDTA) as a solvent. To obtain a homogenous solution, it was heated in a microwave oven for about 1minute. In the fume hood, 10,000X (5μl) of GR Green fluorescent dye (Inqaba biotech South Africa) were added with vigorous shaking and was allowed to cool. It was poured in gel casting tray after cooling down to about 40 °C. The prepared gel was placed in the electrophoresis tank and x1 TBE buffer was added to submerge the gel. The extracted genomic DNA samples (5μl) mixed with 1μl of 6x DNA loading dye from Thermo Scientific was loaded in the wells of the gel accordingly as well as the step-ladder DNA (Thermo Scientific O' Gene Ruler, 1kb plus). The set up was covered and connected to the power pack and was run at 55volts for 45 minutes, after which it was visualized using Alpha Innotechdocumentationtion system model 201103 (Taiwan). This process was carried out for each of the extraction methods optimized [23].
Concentration of DNA ( μl /ml ) =
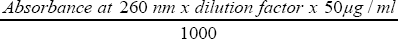
E stimati on of the quality and quantity of the extracted genomic DNA by UV spectrophotometry: For spectrophotometric analysis, 2μl of DNA samples was diluted to 100μl with T.E. buffer and 100μl of the blank solution were prepared. The spectrophotometer readings was recorded, the blank sample was read in the machine first, the DNA samples diluted in T. E. buffer was recorded at 260nm/280nm using Spectophotometry (UV absorbance) at 260nm and 280nm, using U.V Spectophotometer (Lab Kit model ST-UV 755B). DNA concentration was calculated using absorbance values at 260 nm using the following formula [23].
RAPD-PCR
A set of eleven (11) primers obtained from Inqaba Biotech (South Africa) was used for RAPD-PCR. The primer designation and sequence are presented in Table 1.

(Inqababiotecch)
Preparation of stock solutions for primers: This was prepared according to manufacturer's instruction. Stock solutions of primers were prepared in a sterile T. E. buffer solution (10mM TrisHCl, pH 7.5 to 8.0, 1mM EDTA).
Preparation of T E buffer: One times (1X) TE buffer solution consists of 10mMTris, adjusted to pH 7.5 with HCl and one (1) mM EDTA. 1ml of TrisHCl was measured from an already prepared 1M stock solution of TrisHCl and 0.2mls of EDTA was measured from 0.5M stock solution of EDTA. This solution was made up to 100mls with distilled water, sterilized and stored at room temperature.
Constitution of primers: Lypholisedoligos were centrifuged to obtain pellets that might be displaced from the bottom of the tube during shipment. Primer stock solution was prepared (100μM) with sterile T E buffer solution. Working solutions were diluted from the stock solution with sterile nuclease free water to prevent inhibition of enzymatic reactions and degradation of the nucleic acids by nucleases. From the stock solution, 25μl (15μM concentration), was divided into smaller aliquots for long term storage to avoid frequent freeze thaw cycles and accidental contamination.
Preparation of working solutions for primers: Fifteen (15) μM concentrations of primers for working solution was prepared from the stock solution. Microcentifuge tubes were labeled for working solutions appropriately. The tubes were disinfected with UV light in equipped laminar flow hood for 15min. Eighty - five (85) μl of T.E. buffer were added to each tube. Twenty five (25) μl of the 100μM primer stock solution were added to each tube. It was vortexed for 30 seconds at 3,000rpm. Tubes were sealed with parafilm and placed in -20 °C freezer. Eleven (11) tubes to be used within the month were kept in -20 °C, while the rest was stored in a -80 °C freezer.
Procedure for RAPD-PCR: All reagents needed for RAPD- PCR were arranged in a freshly filled ice bucket. They were allowed to thaw completely before setting up the reaction. The RAPD-PCR Program/cycling conditions inputted into the PCR machine (Peltier Based Thermacycler Labkits PCR-MGL- 96+/Y) are presented in Table 2.

RAPD-PCR reaction mixture: PCR tubes were labeled accordingly with an ethanol resistant marker. A mastermix was prepared on ice in 6 eppendorf 1.5ml tubes for the different primers (OPB 01, OPB 02, OPD 01, OPF 01, OPF 2, and OPJ 01). The PCR reagents were pippetted into the tube in the following order; sterile water, BSA, Primer, and Taq polymerase Master mix. It was mixed gently by pipetting up and down for at least 20 times. A 96 well plate was placed in the ice bucket as a holder for the 0.2ml thin walled PCR tubes. Three (3) μl of the DNA template was added into the PCR tubes. Twenty two (22) μl of the mastermix cocktail was gently added to the DNA template in the PCR tubes to make up the total volume of reaction to 25ul. The various components used for RAPD-PCR reaction are presented in Table 2. The tubes were placed in the PCR machine and covered. The RAPD-PCR program was run using the cycling conditions presented in Table 3. After the PCR programme was finished the tubes were removed from the thermocycler and stored at 4 °C in the refrigerator. This procedure was repeated for primers OPJ 02, OPA 01, OPA 02, 0PL 01 and OPL 02.

PCR agarose gel electrophoresis: One percent (1.5%) agarose gel was prepared by weighing accurately 0.75g of agarose powder, which was added to 50mls of 1X TBE (Tris boric EDTA) buffer; it was dissolved in a microwave oven for 2min to obtain a clear solution. GR Green (fluorescent dye) was added to the solution with vigorous shaking and allowed to cool for 40 °C. The gel was poured into the gel casting tray and was allowed to set for about 20min. The prepared gel was placed in the electrophoresis tank and 1X TBE buffer was added to cover the gel. Five (5) μl of the RAPD - PCR product was loaded into wells on the agarose gel. Two (2) μl of the 10, 000bp and 1000bp molecular weight marker (Fermentas) were also loaded in the first and last well. Agarose gel electrophoresis was run for 45 min at 55volts. It was then visualized with a gel documentation system and photographed.
Analysis of data: The RAPD profile was scored for the presence, (represented as 1) or absence (represented as 0) of bands of various molecular weight sizes in the form of binary matrix. The molecular data collected were subjected to Numerical Taxonomy System of Statistic (NTSYS). In NTSYS analysis, the similarity indices between pairs of genotypes were calculated for the combination of the data from the molecular studies by selecting for Qualitative Analysis (SimQual) using method of Jaccard (1908). The similarity matrix is used for clustering of the genotypes based on Unweighted Pair Clustering Group Arithmetic Average (UPGMA). Which does not consider the joint absence of the character as an indication of similarity by selecting Sequential Hierarchical Numeric option (SHAN) in clustering and the clustering was used to generate dendrogram by selecting tree plot option.
The similarity index of Jaccard between plant i and j is given by
Sij = a / ( a + b + c )
Dij =1 - Sij
Where a , is the number of characters present in the plants i and j , b is the number of characters present in i not in j and C is the number of characters present in j and not in i and d is the distant coefficient.
Results and Discussion
DNA extraction
The DNA picture of the plant sample showed that most of the DNA band is close to the neck of the well. The DNA band is heavy and because of the heaviness, they remain close to the neck of the well. The Ladder used here is a 10kb. The herbal DNA band is also heavy and close to the neck of the well. There is some smearing and sheared DNA band, though there is the presence of some other constituent of the herbal sample (phenol).
Quantity and quality analysis result of genomic DNA extracted
The quantity and quality of genomic DNA extracted from plant sample showed that the absorbance is better. While, the quantity and quality of genomic DNA extracted from herbal sample was not so good and the adsorbance is a little low, as such it showed poor result.
PCR Picture of the different primer on both the herbal and plant sample
The PCR Picture of Primer OPL 01 plant and herbal Sample with sequence GGCATGACCT and those of Primer OPA 01 herbal and plant Sample with sequence CAGGCCCTTC, showed that there was polymorphism across the different plant sample and there location. They were quite different from those of the plant sample. The band observed were scattered as such two different ladders were employed 10kb and 1kb. The band observed in sample 2 to 5 had some smear.


The PCR Picture of Primer OPJ 01 plant and herbal sample with sequence CCCGGCATAA as presented in Plate 1 & 2, showed polymorphism across the sample and there location but the plant sample pattern was different from those of the herbal sample of the same primer.


Primer OPA 02 herbal and plant Sample with sequence TGCCGAGCTG Plate 3 & 4 showed that there was polymorphism across the different plant and herbal sample location but they were quite different from those of the herbal sample. The band observed were also scattered but closely related between 1000 - 1500 band weights for plant sample.


PCR Picture of Primer OPB 01 plant and herbal Sample with sequence GTTTCGCTCC as presented in Plate 5 &6 showed that there was polymorphism across the different herbal sample. The band observed was almost similar in their pattern.
PCR Picture of Primer OPF 01 plant and herbal sample with sequence GAGGATCCCT showed that there was also polymorphism across the different plant sample. The band observed was scattered and though there was a sample with a smear.
Scoring of the Different Primers for both the Herbal and Plant Sample
Image profiles of the banding patterns as obtained in the PCR gel picture were recorded and molecular weight of each band as determined by NTSYS Analyst software showed polymorphism as well as monomorphic appearance. Most intense monomorphic band from each accession with each primer was used as reference to calibrate different lanes for the amounts of DNA present (Table 4). When the band was not monomorphic, the band with maximum frequency in each accession was used for calibration. In each lane, bands with at least 10% intensity were scored present of the monomorphic reference band within the same lane [24].
Table 4 showed the scoring of the herbal sample primer OPL 01 with sequence (1) GGCATGACCT, OPA 01 with sequence (2) CAGGCCCTTC, OPJ 01 with sequence (3) CCCGGCATAA, OPA 2 with sequence (4) TGCCGAGCTG and OPB 01 with sequence (5) GTTTCGCTCC. OPL 01 has two polymorphic band at 1000 and 900 across the different location. While at band weight 800 and 700 the sample was unique. OPA 01 also had two polymorphic bands but at 900 and 800 and it was monomorphic at band weight 1000. Primer OPJ 01 had two monomorphic band weight at 1000 and 900 and polymorphic 800. OPA 02 had three polymorphic band weight at 1500, 900 and 800 but monomorphic at 1000 band weight. OPB 01 showed three polymorphic bands at 1000, 800 and 700 while it was unique at 600 and 500 but monomorphic 900.


Table 5 showed scoring of primers OPL 01 plant sample (1) GGCATGACCT, OPA 01 plant sample (2) CAGGCCCTTC, OPJ 01 plant sample (3) CCCGGCATAA, OPA 02 plant sample (4) TGCCGAGCTG, OPB 01 plant sample (5) GTTTCGCTCC. OPL 01 was polymorphic at 1500 and 1000, but unique at 700. OPA 01 result also shows polymorphism at 1000 and 700 band weight, it also had one monomorphic band weight at 900. OPJ 01 has two polymorphic (1500 and 700) and unique (800 and 600) band weights and only one monomorphic band weight at 1000. OPA 02 had three polymorphic band weights at 1500, 1000, 900 and unique at 800. OPB 01 was polymorphic at 1000, 900, 800 and 600. Though, it was unique at 700 and 500 band weight.
Percentage Polymorphism of Primers
Table 6 showed the percentage polymorphism of the different primers across the plant sample. It was observed to be 66.6% for OPA 01 and 80% for OPJ 01 wish implies that the polymorphism did not cut across the plant sample completely. While, OPL 01, OPA 02 and OPB 01 had 100% polymorphism.


Table 7 also showed the percentage polymorphism of primers for herbal sample. It was 100% in OPL 01 primer only. While OPA 01, OPJ 0, OPA 02 and OPB 01 had 66.6%, 33.3 %, 75% and 83.5% respectively.
Numerical Taxonomy System of Statistic Analysis
Numerical Taxonomy System of Statistic (NTSYS) analysis showed only 37.5% relatedness in the 12 herbal sample and 12 plant sample collections, which is deemed to be useful in authentication of herbal product and identification of medicinal plant material.
The output of the data from the NTSYS generated a dendrogram showing the genetic diversity among the samples under study as shown in Figure 1. The cluster generated from the dendogram contained two distinct clusters. Group 1 contained only one sample while the rest accessions were clustered in group 2. Group 1 contained Garaku Nasarawa plant sample at 0.63 similarity index. Group 2 has two separate clusters that grouped the remaining twenty-three both herbal samples and plant samples. The first cluster of group 2 contains 3 subgroups that had 11 herbal samples with Zuba FCT plant sample and Auta Nasarawa plant sample in one sub group, while Giri FCT plant sample and kubwa FCT plant sample clustered together, the implication is that they have similar genetic traits at that coefficient. Kaduna road plant sample and New Karu Nasarawa sample clustered from the other accessions leaving Apo FCT plant sample, Keffi Nasarawa plant sample clustered with Uke Nasarawa plant sample, it means that at 0.63 similarity index both of them have the same genetic composition hence there is no difference among them.

The other clusters in group 2 consist of three major clusters that made up 100% of the herbal samples under study. The first subgroup had four herbal samples obtained from Zuba, Kubwa, Giri all from FCT with Auta from Nasarawa herbal sample. This showed they have the similar composition. Although New Karu Nasarawa herbal sample clustered with them but with little degree of genetic differences, Nyanyan FCT herbal sample stands separately from Kaduna Road herbal sample and Keffi Nasarawa herbal sample. At 0.63 similarity index herbal samples from Garaku Nasarawa has the same content in terms of ingredient. Orozo FCT herbal sample and Apo FCT herbal sample made up the remaining accessions in the third sub-cluster.
Traditional medicinal systems uses whole plants or their parts for combating a great number of disorders. Hence, maintaining the purity and quality of herbal drugs are necessary to identify the substitutes and adulterants. Medicinal plants constitute an effective source of traditional and modern medicine. Adulterations and substitutions are common in herbal material trade and medicinal plants as observed in the result. Adulteration in market samples is one of the greatest drawbacks in promotion of herbal products which could be the reason for the 37.5% relatedness obtained in the phylogenetic tree. Due to this adulteration and altered efficacy, the faith in crude drug promotion has declined [25]. The RAPD, PCR based assay described here is fast, reliable, and easy to conduct in any laboratory. It can be carried out in very short period using unknown genomic DNA from any developmental stages and body parts of herbs [26]. Despite close identity as observed in the PCR analysis and Picture, a great deal of polymorphism was observed among the accessions. Khaya senegalensis and Azadirachta indica also showing significant genetic variations with each other.
Conclusion
From the phylogenetic tree generated among the 24 samples based on PCR product, it was observed that the relatedness which stand at 37.5% is an appreciable relationship compare to many others. This showed that they contain both Khaya senegalensis and Azardirachta indica as they claim though; the quantity is what is now in contempt. This is simply because 37.5% of a result is not good enough for the loud claim of the ability of both Khaya senegalensis and Azardirachta indica to treat Diabetes.
The percentage of the herbal sample is very low and as such cannot be said to have such herbal potency to cure diabetes as claimed. Though this is not to say other content of the sample do not have such potency as such statement is subject to research. The herbal sample also contains a lot of mixture that made it difficult to conclude its potency.
Form the DNA extraction process, a lot of phenolic substance was observed as the DNA was cloudy in some case. The quality of the DNA was also not good as observed under the Gell Documentation System. Development of RAPD markers that can correlate DNA fingerprinting data with quantity of selected phytochemical markers associated with a specific medicinal herb would have extensive applications in quality control of herbal product and also on their raw materials. Other herbal claims by this local herbs seller should also be research in and around Africa because there are a lot of yet to be discovered genuine herbal combinations that treat many ailments. Molecular markers should always be employed as a qualitative/quantitative diagnostic tool for identification of medicinal herbs from harvesting to finished product.
References
- Hart BL (2005) The evolution of herbal medicine: Behavioural perspectives. Animal Behav 70(5): 975-989.
- Halberstein RA (2005) Medicinal plants: historical and cross-cultural usage patterns. Ann Epidemiol 15(9): 686-699.
- Huffman MA (2001) Self-medicative behavior in the African great apes: an evolutionary perspective into the origins of human traditional medicine. Bioscience 51(8): 651-661.
- Krief S, Hladik CM, Haxaire C (2005) Ethnomedicinal and bioactive properties of plants ingested by wild chimpanzees in Uganda. J Ethnopharmacol 101(1-3): 1-15.
- Ernst E (2005) The efficacy of herbal medicine: an overview. Fundam Clin Pharmacol 19(4): 405-409.
- Cardellinaj HI (2002) Challenges and opportunities confronting the botanical dietary supplement industry. J Nat Prod 65(7): 1073-1078.
- Ivorra MD, Paya M, Villar A (1989) A review of natural products and plants as potential antidiabetic drugs. J Ethnopharmacol 27(3): 248275.
- Bnouham M, Mekhfi H, Legssyer A, Ziyyat A (2002) Medicinal plants used in the treatment of diabetes in Morocco. Int J Diabetes & Metabolism 10: 1-33.
- Al-Rowais NA (2002) Herbal medicine in the treatment of diabetes mellitus. Saudi Med J 23(11): 1327-1331.
- Dey L, Anoja SA, Yuan CS (2002) Alternative therapies for type 2 diabetes. Altern Med Rev 7(1): 45-58.
- Matteucci E, Giampietro O (2000) Oxidative stress in families of type I diabetic patients. Diabetes Care 23(8): 1182-1186.
- De Bie S, Ketner P, Geerling P, Geerling C (1998) Woody plant phenology in the West Africa savanna. Journal of Biogeography 25(5): 883-900.
- Singh A, Negi MS, Rajagopal J, Bhatia S, Tomar UK, et al. (1999) Assessment of genetic diversity in Azadirachta indica using AFLP markers. Theoretical and Applied Genetics 99(1-2): 272-279.
- Kundu SK (2000) Evaluation of provenance variation on early growth and survival of neem (Azadirachta indica) in Bangladesh and India. Journal of Tropical Forest Science 12(3): 509-523.
- Morell MK, Peakall R, Appels R, Preston LR, Lloyd HL (1995) DNA profiling techniques for plant variety identification. Australian Journal of Experimental Agriculture 35(6): 807-819.
- Welsh J, McClelland M (1990) Fingerprinting genomes using PCR with arbitrary primers. Nucleic Acids Res 18(24): 7213-7218.
- Williams JGK, Kubelik AR, Livak KJ, Rafal-ski JA, Tingey SV (1990) DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res 18(22): 6531-6535.
- Gunther H (2010) New aspect of DNA-based authentication of Chinese medicinal plant by molecular biological techniques. Planta Med 76(17):1963-1974.
- Akwe VL, Binbol NL, Samaila KL, Marcus ND (2007) Geographical and perspective of Nasarawa State. Onaivi Printing and Publishing Company Limited, Keffi, Nigeria, p. 30.
- Ochuko A (2013) Evaluation of Environmental Noise Pollutionin Abuja, the Capital City of Nigeria. International Journal of RRAS 14(2): 24.
- Vengadesh PN, Xinxu Z, Yuki M, Fumio I, Fengping W (2016) A modified SDS-Based DNA extraction method for high quality environmental DNA from seafloor environment. Front Microbiol 7: 986-992.
- Dellaporta SL, Wood J, Hicks JB (1983) A plant DNA mini-preparation: Version II, Journal of Plant Molecular Biology 1: 19-21.
- Charles O, Salisu A, Gabriel M, Ogechi N, Veronica E, et al. (2015) Extraction of good quality genomic DNA from dry woody mushroom samples for molecular analysis: A case study of Ganoderma lucidum. Journal of Translational Medicine and Biotechnology 3(5): 1-9.
- Anjum G, Ashish G, Vikas H, Rajesh D (2003) RAPD Patterns of some important medicinal plant and their substitutes used in ayurveda to identify the genetic variations. International Journal of Pharmacy and Pharmaceutical Sciences 5(1): 239-241.
- Dubey NK, Kumar R, Tripathi P (2004) Global promotion of herbal medicine: India's opportunity. Current Science 86(1): 37-41.
- Kethidi DR, Roden DB, Ladd TR, Krell PJ, Retnakaran A, et al. (2003) Development of SCAR markers for the DNA-based detection of the Asian long-horned beetle; Anoplophoragh bripennis (Motschulsky). Arch Insect Biochem Physiol 52(4): 193-204.















