




Variation of Prokaryotic and Fungal Soil Communities across a Vegetative Transect
Sarah E Shawver1, Jeff A Brady2 and Donald G McGahan1,2
1Department of Wildlife, Sustainability and Ecosystem Sciences, Tarleton State University, USA
2Texas A&M AgriLife Research, USA
Submission: March 09,2018; Published: April 11, 2018
*Corresponding author: Donald G McGahan, Department of Wildlife, Sustainability, and Ecosystem Sciences, Tarleton State University, Box T-0050, Stephenville, TX 76402, USA, Email: MCGAHAN@tarleton.edu
How to cite this article: Shawver S E, J A Brady, D G McGahan. Variation of Prokaryotic and Fungal Soil Communities across a Vegetative Transect. Agri Res & Tech: Open Access J. 2018; 15(3): 555953. DOI:10.19080/ARTOAJ.2018.15.555953.
Abstract
Soil microbial communities depend on pH, texture, and available nutrients. Ashe juniper (Juniperus ashei) is encroaching on live oak (Quercus virginiana) savannas. Litter differences may alter soil microbial communities and therefore the ability of the soil ecosystem to cycle nutrients. The objective was to determine whether soil microbial communities differ under the canopies of Ashe juniper and live oak. At each of three transects we collected four surface soil samples under live oak canopy, overlapping canopies of live oak and Ashe juniper, under Ashe juniper canopy, and in grass away from the trees. We characterized soils by pH, texture, calcium carbonate equivalence, total nitrogen, and total carbon. Microbial communities were analyzed using massively parallel DNA sequencing and analyzed within QIIME using Kruskal-Wallis ANOVA, Mann-Whitney U, and Spearman’s rank correlations. Prokaryotic communities differed along the transect, while fungal communities did not. Soil pH increased from live oak to grass. Trends in taxa were mostly associated with vegetation, pH, and CCE. Many prokaryotic genera associated with soil properties were important nutrient cyclers. Many fungal genera associated with pH and vegetation were mycorrhizal fungi, suggesting a plant-specific relationship.
Keywords: 16S; ITS; Sequencing; Juniperus; Quercus; Vegetation; Rangeland; Soil health
Introduction
Soil microbial communities are affected by a number of biotic and abiotic factors including pH, nitrogen (N), carbon (C), and vegetation. Soil pH is one of the primary influences on microbial communities. Soil chemical properties, especially pH, rather than spatial effect, land use, climate, or soil physical properties, explain the largest amount of variance in microbial communities [1]. Soil pH impacts microbial and fungal diversity, but temperature and latitude do not, and greater diversity is found where soil pH is more basic [2]. Additionally, Arbuscular mycorrhizal communities are dependent on pH, carbonate content, base saturation, and phosphorus [3].
In addition to pH, nutrient availability impacts microbial communities. Microbial biomass is directly related to soil organic carbon content [4,5]. Not surprisingly, spatial distribution of denitrifying organisms within a field is related to nitrates, dissolved organic C, and dissolved organic N [6].
While pH and nutrients can individually impact distribution and abundance of microbial communities, these factors can also have a synergistic effect. Soil pH affects the availability of nutrients. Wetter climates tend toward greater base cation leaching, acidity, and fungi:bacteria ratios [7]. With less moisture available for leaching, soil pH and base cation content are typically greater.
Vegetation also impacts soil microbial communities. Globally, microbial communities are impacted by biome and belowground plant net primary production [8] while soil microbial community composition varies with ecosystems [2]. For example, ratios of fungi:bacteria are lower in grasslands due to differences in pH and moisture. While both plants and soils impact soil microbial communities [5], grassland type plays a major role in explaining variability in soil microbial communities of grasslands [9]. When considering both biotic and abiotic habitats, the predictive fit of models in grazing pastures improves when plant traits are included [7], and microbial communities can be quite plant- specific [10]. Given that plants and soil microbes are intricately related, we would expect that changing one would alter the other. Microbial communities can be shaped by the presence of species-specific root exudates, which can provide C and growth factors for auxotrophic organisms [11].
In Texas, Ashe juniper (Juniperus ashei) has been overtaking savannas previously dominated by live oak (Quercus virginiana). This is a major management concern for water-limited Texas rangelands, because interception and loss of precipitation by Ashe Juniper canopies is among the highest of the dominant vegetation types across Texas rangelands [12,13]. Ashe juniper has an advantage over live oak due to greater leaf area for a similar sized tree [14]. Similar to grassland type influences mentioned previously, the different litter inputs between Ashe juniper (coniferous) and live oak (deciduous) may support different microbial communities. In arid climates, resource islands created by shrubs influence both microbial abundance and community structure [15-17]. In semi-arid climates, the presence of woody species encroaching in a grassy landscape could also lead to variation of soil microbial communities.
The encroachment of woody vegetation on a historically grassy landscape could indicate ecosystem changes below ground, as well as above, since the soil microbial community is subject not only to exudates from the vegetation roots but also litter, and because soils under trees tend toward lower pH. Both prokaryotic and fungal communities would likely be altered as a result of the change in fungi: bacteria ratios between forests and grasslands [8] and with moisture [7]. The aggregate ability of the microbial community to cycle nutrients and impact soil organic matter (SOM), and therefore C turnover, also may change.
Litter input chemistry is influential on microbe-driven soil processes such as humification and N mineralization [18]. Oak species are allelopathic, and able to hinder plant growth and germination of acorns underneath the canopy drip line via root and leaf allelopathy, as well as root competition [19]. Tannins have been identified as the compounds most likely responsible for allelopathy [20]. However, soil microbes break down allelopathic chemicals, which likely allows other plants to grow in and around oaks [20,21].
Determining if Ashe juniper supports a principally different microbial community might provide a window to controlling savanna invasion. Identifying that a principally different microbial community is supported in the presence of Ashe juniper might lead to questioning if Ashe juniper is simply immune to the allelopathic compounds produced by live oaks, or if establishment of Ashe juniper at the ecotone between live oak and grassland allows a microbial community under the Ashe juniper canopy an advantage in degrading the live oak allelopathic compounds and thereby lead to enhanced competition for resources under the now shared canopy. No research has yet examined the potential allelopathic effect of Quercus sp. on Juniperus sp.
The primary objectives of this study were to determine whether soil prokaryotic and fungal communities change along a vegetative transect of grass, Ashe juniper, live oak, and identify any specific taxon for future investigations. We predicted that the composition of fungal and prokaryotic communities would change along the vegetative transect, and that soil properties, such as pH, calcium carbonate equivalence (CCE), N, and C, would be associated with the change. Without these initial characterizations of the microbial community, nutrient cycling impacts on woody species invasions such as Ashe juniper into savanna range lands will remain stunted. With a better understanding of the soil below ground savanna rangeland, soil health and potential consequences for rangeland productivity might be better understood.
Methods
Study area
The study site was located in Erath County, Texas USA (N32° 12.264' W98° 05.515'). The juniper and live oak association typically occurs on a limestone shelf. These soils tend to be shallow and have high levels of carbonates. Three transects of live oaks, Ashe juniper, and grass were identified. All transects were from areas mapped as Maloterre consociation (loamy, carbonatic, thermic Lithic Ustorthent) [22].
The transects consisted of 4 locations: under the live oak canopy, under the area that live oak and Ashe juniper overlap, under the Ashe juniper canopy, and in the grass with no tree canopy. For each location along the transect, one pit was excavated to determine the depth of the surface horizon. The surface horizon was sampled by aggregating samples from across the excavation at three additional locations within 2m of the first sample excavation, ensuring that all samples were under the appropriate vegetation canopy, for a total of four aggregated samples at each of the four points along the three transects. For all soils, only bulk soil was analyzed. Samples were air-dried, carbonate masses were removed by forceps, and the soil was ground to pass through a 2 mm sieve.
Soil characterization
Soil pH was determined by 1:1 soil:water using an Accument Research AR20 pH meter (Fisher Scientific International, Pittsburgh, Pennsylvania; [23]). Soil texture was determined by the pipette method after chemical dispersion with sodium hexametaphosphate [23,24]. CCE was determined using a modified method [23,25]. Total N and total carbon (TC) were determined using the Dumas method [23,26] dry combustion as a percentage (Elementar Vario Macro CN analyzer, Hanau, Germany). Organic carbon (OC) was calculated by subtracting CCE carbon from TC.
Microbial community composition
Deoxyribonucleic acid (DNA) was extracted using 300 mg subsamples of soil following the protocol of QIAGEN DNeasy PowerSoil kit. Prokaryotic 16S amplicons were generated using primers 519F (5'-CAGCMGCCGCGGTAA-3') and 785R (5'-GACTACHVGGGTATCTAATCC-3') that amplified the V4 region [2 7,28]. Fungal amplicons were generated using primers (5'-GTGAATCATCGAATCTTTG-3') and (5'-TCCTCCGCTTATTGATATGC-3') that amplified the fungal ITS2 region. The concentration of DNA was determined using a Qubit® 2.0 fluorometer with a Qubit® dsDNA HS Assay Kit according to manufacturer's instructions (Life Technologies, Carlsbad, CA). The DNA was normalized from each sample to a concentration of 5ng/μl before subsequent analysis.
Raw sequencing reads were processed with a combination of QIIME [29], USEARCH [30], and custom python scripts. UCLUST [30] was used to pick referenced-based Operational Taxonomic Units (OTUs) at 97% similarity. A representative set of prokaryotic sequences was compared to the Greengenes 13.8 reference sequence database [31] to assign taxonomy. A representative set of fungal sequences was compared to the UNITE 7.0 fungal reference sequence database [32] to assign taxonomy. OTUs composed of only one sequence were removed. The sequencing dataset was rarified to an equal sequencing depth by randomly subsampling sequences from each sample without replacement in order to provide even measures of microbial alpha- and beta- diversity and to have equal sequencing depth for the production of all figures, tables, and statistical analyses.
Statistical Analysis
Prokaryotic and fungal datasets were analyzed separately. Diversity was calculated using unweighted unifrac distance matrices [33]. OTU diversity was measured using Shannon diversity Index and OTU richness. Differences in microbial community diversity were statistically analyzed with Kruskal- Wallis ANOVA and Spearman's rank correlation using QIIME [34,35]. Bray-Curtis distance was used to ordinate microbial community data with Principle Coordinates Analysis (PCoA). Differences in communities by vegetation were tested with PERMANOVA using the adonis function and betadisp of the package Vegan in R [36]. Mann-Whitney U tests were used to conduct direct comparisons between two conditions or groups. ANOVAs were used to test for significant differences in the soil properties, and the range was determined with average and standard deviation [37]. Bonferroni-adjusted p-values were used in all genomic statistical analyses.
Results And Discussion
Soil properties
Soil properties were not all similar along the transects (Table 1). Soil pH and CCE were associated with vegetation, but not N, TC, and OC. Soil pH was highest in the grass and lowest under the oak canopy Soil pH is typically higher in prairies and grasslands than forested ecosystems [38]. Despite the patchy habitat and similar soil forming factors, the pattern held true. Increased additions of fulvic organic acids under deciduous oak canopies are likely responsible for reduced pH at these sites [20]. CCE was also highest in the grass, perhaps, in part, enhanced by drier conditions away from the shade provided by trees. However, the overlapping canopy of oak and juniper had the lowest CCE, even though there was no statistical difference between the oak and the juniper soil CCE.

We expected soil N and OC variation along the vegetation Vegetation influence on prokaryotic communities transect due to the change in organic inputs from deciduous and coniferous trees and grasses as OC and total N tend to be lower in forests than prairies [38]. Though this study did not detect a significant difference, it should be noted that the OC content of samples was variable.
Prokaryotic communities
A total of 47 samples were sequenced with 1,628,504 sequence reads remaining in the dataset following quality filtering measures. There were 33,927 ± 30,180 sequences per sample and samples were rarified to 3,031 sequences per sample for the purpose of comparison. After OTUs with only one sequence were removed, there were 6,780 OTUs. The most abundant phyla were Acidobacteria (23%), Proteobacteria (19%), Actinobacteria (16%), Crenarchaeota (11%), and Plancto mycetes (7%). Twelve OTUs were present in every sample.
Vegetation influence on prokaryotic communities

We anticipated that microbial communities may change gradually along the transition from live oak to Ashe juniper to grass. The community composition along the transect was not uniform. While samples taken from grass and under the juniper canopy have communities very similar to each other, these are distinct from communities from samples taken under the oak and the combined oak and juniper canopy. The soil under the combined canopy has a unique composition, and has a more even distribution of phyla (Figure 1).



OTU diversity, as measured by Shannon Index and OTU richness was lower in live oak and juniper-oak than in Ashe juniper and grass (p<0.05) (Table 2). We expected the area of overlapping canopies, juniper-oak, might have a microbial community reflecting both Ashe juniper and live oak communities. Surprisingly, juniper-oak diversity was significantly lower than Ashe juniper, and more similar to live oak. The lower diversity under live oak could be due to allelopathic compounds suppressing some organisms. We interpret this lower diversity under the juniper-oak canopy to mean that allelopathic compounds, such as tannins, from oak exudates and leaf litter might remain a strong influence in the soil under the overlapping canopy, reducing the diversity of the microbial community [20]. Additionally, looking at beta diversity with PCoA, we expected to see samples cluster by vegetation, and juniper-oak samples to be similar to both Ashe juniper and live oak samples. However, using PERMANOVA, juniper-oak and live oak samples were significantly different from Ashe juniper and grass samples (p<0.05) (Figure 2). Again, this is interpreted as an influence exerted by the live oak. Additionally, the microbial communities were more variable in grass and Ashe juniper communities than live oak and juniper-oak (p<0.05) (Table 3).
Four phyla: TM7, Firmicutes, Armatimonadetes, and Gemmatimonadetes were associated with vegetation based on Kurskal-Wallis ANOVA (p<0.05). The change in relative abundance was not uniform among the phyla. TM7 and Armatimonadetes decreased from grass to live oak, while Firmicutes was highest under Juniper-oak, based on Mann-Whitney U comparisons. Gemmatimonadetes decreased from Ashe juniper to juniper- oak, but was not different between grass and live oak (Table 4). With all four phyla, relative abundance between grass and Ashe juniper was similar, and the relative abundance between juniper- oak and live oak were similar. While live oak seems to exhibit an influence that generally persists under the shared juniper- oak canopy, we interpret greater abundance of Firmicutes and decrease in Gemmatimonadetes abundance under the shared juniper-oak canopy to mean that juniper encroachment may impact some prokaryotes, but not all.

When considering prokaryotic genera, thirty-four were significantly associated with vegetation based on Kurskal- Wallis ANOVA (p <0.05). Many of those genera are important biogeochemical cyclers with mixed trends. For example, Variovorax (Proteobacteria phyla), a nitrate-reducer [39], decreased in abundance from grass to live oak, while Pseudomonas (Proteobacteria phyla), a denitrifier [40], increased from grass to live oak. While we did not find significant variation in total N along the vegetation transect, the change in abundance of these genera may indicate a change in the dominant forms of N along the transect. Although characterizing the relative amounts of the various forms of N was beyond the scope of this study, this study suggests that characterization of N forms is a likely line of inquiry for future transects studies on these semi-arid rangelands. We can deduce from these trends that moisture shifts along the transect are reflected in the relative shift in community organisms. Since denitrification is an anaerobic process, it follows that the moisture status from the more arid grass to the shadier and lower evapotranspiration live oak site fits the community trend, and nitrate reduction, an aerobic process, represents a reciprocal trend.
Soil pH and C influence on prokaryotic communities
The relative abundance of five phyla: Armatimonadetes, FBP, Firmicutes, TM7, and Cyanobacteria were significantly associated with pH based on Spearman's rank correlation, with mixed trends (p<0.05). Armatimonadetes, FBP, and TM7 had a positive relationship with pH, while Firmicutes and Cyanobacteria had a negative relationship with pH. Thirty genera were significantly associated with pH based on Spearman's rank correlation (p<0.05), 17 of which were also associated with vegetation, corresponding with the shift in pH along the transect.
Four phyla: Crenarchaeota, Gemmatimonadetes, Nitrospirae, and Fibrobacteres were significantly associated with CCE based on Spearman's rank correlation (p<0.05). All but Nitrospirae had a positive relationship with CCE. At a lower taxonomic level, twenty-seven genera, including Candidatus nitrososphaera, an ammonia-oxidizing organism [41], and Flavobacterium, were significantly associated with CCE based on Spearman's rank correlation (p<0.05). Flavobacterium is involved in C cycling as it has the potential to break down poly-aromatic hydrocarbons [42].
Many genera were significantly associated with multiple parameters. For example, there were prokaryotes that were significant with vegetation, pH, and CCE. We have shown that both pH and CCE increased from live oak to grass, likely resulting from the change in leaf litter deposition and amount, moisture content, and metabolism differences. As pH is inextricably related to CCE, it is intuitive that abundance of responsive taxa would be highly overlapped between the two parameters, and related to vegetation. Fifteen genera were associated with both vegetation and pH. Three genera were associated with both vegetation and CCE, and two genera were associated with all three parameters. Despite the large overlap, there were still 14 genera that were only associated with vegetation, indicating that plant specific exudates and leaf litter composition may have a unique influence on soil prokaryotic communities.
Fungal communities
A total of 40 samples were sequenced with a total of 859,945 sequence reads remaining in the dataset following quality filtering measures. There were 17,916±17.059 sequences per sample and samples were rarified to 1,478 sequences per sample for the purpose of comparison. After OTUs with only one sequence were removed, there were 1,176 OTUs. No OTUs were present in more than 65% of samples. Unlike the prokaryotes, two phyla made up 99% of all sequences (Ascomycota, 76%, and Basidiomycota, 23%).
Vegetation influence on fungal communities
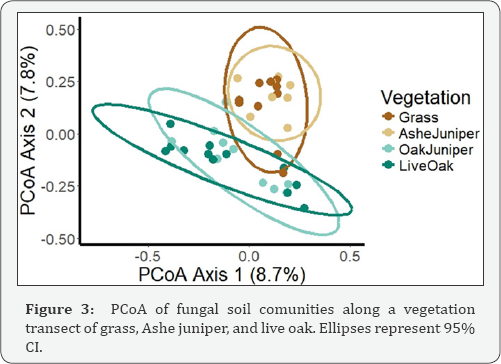
We expected fungi to be more diverse under woody canopy ecosystems than grasslands [8]. Along the transect, we expected the grass and, to some extent, the Ashe juniper to act as a grassland and the juniper-oak and live oak to act as a forest. Even though the sampling areas were relatively small, studies have shown that small patches of vegetation can act as resource islands and impact soil microbial communities [1517]. Therefore, we predicted a change in fungal diversity along the transition from live oak to Ashe juniper to grass. Although we did see a change in diversity in prokaryotes, there were no differences in fungal diversity in terms of Shannon Indices or species richness (p>0.05). However, we did detect patterns in community structure through PCoA (Figure 3). Similar to the prokaryotes, the fungal samples clustered by vegetation, with Ashe juniper and grass samples significantly different from Juniper-oak and live oak (Table 3).
Three phyla: Basidiomycota, Ascomycota, and Glomeromycota were significantly associated with vegetation based on Kruskal- Wallis ANOVA (p<0.05) (Figure 4). Ascomycota and Glomeromycota decreased from grass to live oak, and Basidiomycota increased from grass to live oak. Four genera: Alternaria (Ascomycota), an unidentified genus within the Mycosphaerellaceae family (Ascomycota), Rhizophagus (Glomeromycota), and an unidentified genus within the Glomeraceae family (Glomeromycota) were significantly associated with vegetation based on Kruskal-Wallis ANOVA (p<0.05) (Table 5). While Alternaría is a highly diverse genus that is hard to generalize, oak and juniper-oak sites were significantly different from the grass and juniper sites. Rhizophagus and Glomeraceae, both arbuscular mycorrhizae, were highest in grass, suggesting plant-specific interactions. Although previous studies have documented associations of Rhizophagus with both monocot and dicot crops suggesting low plant specificity [43-45]. We believe that in these savannah rangelands future investigations might confirm that these arbuscular mycorrhizae enable grass vigor which is a goal in rangelands Three phyla: Basidiomycota, Ascomycota, and Glomeromycota were significantly associated with pH based on Spearman's rank correlation (p<0.05). Ascomycota and Glomeromycota increased with pH, while Basidiomycota decreased. The changes in relative abundance of these phyla are associated with both pH and vegetation, and show consistent patterns. Ascomycota and Glomeromycota had the greatest relative abundance with high pH and in grass. As grass had the highest pH along the transect, it is unclear whether pH, vegetation, or both impact the abundance of fungal phyla. Six genera: an unidentified genus within Ascomycota, Inocybe, Aureobasidium, Alternaria, and an unidentified genus within Agariaceae (Basidiomycota) were significantly associated with pH Spearman's rank correlation (p <0.05). All genera, except for Inocybe, an ectomycorrhizal fungus, increased with pH [46-53].


One phylum, Glomeromycota, was positively associated with CCE based on Spearman's rank correlation (p<0.05). Three genera: Alternaria, Tuber, and an unidentified genus within Glomeraceae, were significantly associated with CCE based on Spearman's rank correlation (p<0.001). While Tuber was negatively related to CCE, Alternaria and Glomeraceae were positively related.
Conclusion
We predicted that soil microbial communities would change along a vegetative transect of grass, Ashe juniper overlapping canopy, and live oak. As expected, a larger number of prokaryotic OTUs (6,780) were detected than fungal OTUs (1.176). Within the prokaryotes, grass and Ashe juniper had higher diversity and species richness than juniper-oak and live oak, while both prokaryotes and fungi displayed community structures related to plant canopy and soil properties. The grass site is presumed to be drier, and slightly more alkaline. Most essential nutrients are less mobile in more alkaline conditions. Interception and mobilization by a more robust fungal component in the Texas rangeland grass sites might be better characterized to facilitate more informed soil health recommendations. Additionally, future transects in this system should include characterization of available water holding capacity, actual water status seasonally, and availability of micronutrients.
In all comparisons of taxa with vegetation, juniper-oak and live oak were always similar, while Juniper-oak and Ashe juniper were sometimes different, suggesting that the oak may have a stronger influence on the soil microbial community under the overlapping canopy than the juniper. The encroachment of Ashe juniper on live oak savannahs does not appear to affect the relative abundance or diversity of microbial communities underneath the live oak canopy. However, there were differences between live oak and Ashe juniper, suggesting that the belowground community is more variable beyond the limit of the oak canopy
We found that across the relatively short distance of the transect, there was a decrease in pH and calcium carbonate equivalence from grass to live oak. The increase of organic acids from yearly deposits of deciduous leaves under the juniper- oak and live oak likely contributed to the change in soil pH and decreased carbonates. Soil pH and calcium carbonate equivalence were also the main influences on both fungal and prokaryotic communities.
Many phyla and genera of prokaryotic and fungal communities were significantly associated with more than one parameter. As many soil properties are related to one another, such as pH and calcium carbonate equivalence, it is intuitive that we would observe overlapping correlations. However, this overlap was not perfect, suggesting that every soil property has some unique influence on soil microbial communities.
Many of the prokaryotic genera that were associated with vegetation and soil properties were important biogeochemical cyclers. The change in these organisms, particularly those that change with vegetation, could indicate a change in the soil's ability to cycle N and C under an invading plant, such as Ashe juniper. Meanwhile, many of the fungal genera that were associated with vegetation and soil properties indicated a likely difference in mycorrhizal symbioses.
This study suggests that, in addition to soil moisture status, future research should examine the variation of prokaryotes and fungi across other vegetation transects and characterize the different forms of N, OC, and micronutrients. If the Ashe juniper and live oak soil microbial communities have different nutrient cycling capabilities, future studies should examine the impact on biogeochemical cycling and potential consequences for rangeland productivity, carbon sequestration, and soil health. As sequence databases become more complete and our knowledge of soil microorganisms grow, we can gather more knowledge about biogeochemical cycling and the microorganisms that drive it.
Acknowledgements
This work was supported by the USDA National Institute of Food and Agriculture, Hatch project 204995, and Texas A&M AgriLife Research.
References
- Bru D, Ramette A, Saby NPA, Dequiedt S, Ranjard L, et al. (2011) Determinants of the distribution of nitrogen-cycling microbial communities at the landscape scale. International Society for Microbial Ecology 5(3): 532-542.
- Fierer N, Jackson RB (2006) The diversity and biogeography of soil bacterial communities. Proceedings of the National Academy of Science 103(3): 626-631.
- Jansa J, Erb A, Obreholzer H, Smilauer P, Egli S (2014) Soil and geography are more important determinants of indigenous arbuscular mycorrhizal communities than management practices in Swiss agricultural soils. Molecular Ecology 23(8): 2118-2135.
- Fierer N, Allen AS, Schimel JP, Holden PA (2003) Controls on microbial CO2 production: a comparison of surface and subsurface horizons. Global Change Biology 9(9): 1322-1332.
- Garbeva P, van Veen JA, van Elsas JD (2004) Microbial diversity in soil: selection of microbial populations by plant and soil type and implications for disease suppresiveness. Annu Rev Phytopath ol 43: 243-270.
- Enwall K, Throback IN, Stenberg M, Söderström M, Hallin S (2010) Soil resources influence spatial patterns of denitrifying communities at scales compatible with land management. Appl Environ Microbiol 76(7): 2243-2250.
- de Vries FT, Manning P, Tallowin JRB, Mortimer SR, Pilgrim ES, et al. (2012) Abiotic drivers and plant traits explain landscape-scale patterns in soil microbial communities. Ecology Letters 15(11): 1230-1239.
- Fierer N, Strickland MSLD, Bradford MA, Cleveland CC (2009) Global patterns in belowground communities. Ecology Letters 12(11): 12381249.
- Grayston SJ, Griffith GS, Mawdsley JL, Campbell CD, Bardgett RD (2001) Accounting for variability in soil microbial communities of temperate upland grassland ecosystems. Soil Biology and Biochemistry 33(4-5): 533-551.
- Hartmann A, Schmid M, van Tuinen D, Berg G (2009) Plant-driven selection of microbes. Plant and Soil 321(1-2): 235-257.
- Berg G, Smalla K (2009) Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbial Ecology 68(1): 1-13.
- Lyons RK, Owens MK, Machen RV (1998) Juniper biology and management in Texas. Texas Agricultural Extension Service Bulletin 6074: 1-10.
- Thurow TL, Hester JW (1997) Juniper biology and management in Texas. In Juniper Symposium, San Angelo, Texas, USA.
- Owens MK (1996) The role of leaf and canopy-level gas exchange in the replacement of Quercus virginiana (Fagaceae) by Juniperus Ashei (Cupressaceae) in semiarid savannas. American Journal of Botany 83(5): 617-623.
- Bachar A, Ines M, Soares M, Gillnor O (2012) The effect of resources islands on abundance and diversity of bacteria in arid soils. Microbial Ecology 63(3): 694-700.
- Ben-David EA, Zaady E, Sher Y, Nejidat A (2011) Assessment of the spatial distribution of soil microbial communities in patchy arid and semi-arid landscapes of the Negev Desert using combined PLFA and DGGE analyses. Microbial Ecology 76(3): 492-503.
- Smith JL, Halvorson JJ, Bolton H (1994) Spatial relationships of soil microbial biomass and C and N mineralization in a semi-arid shrub- steppe ecosystem. Soil Biology and Biochemistry 26(9): 1151-1159.
- Aber JD, Melillo JM, McClaugherty CA (1990) Predicting long-term patterns of mass loss, nitrogen dynamics, and soil organic matter formation from initial fine litter chemistry in temperate forest ecosystems. Canadian Journal of Botany 68(10): 2201-2208.
- McPherson JK, Thompson GL (1972) Competitive and allelopathic suppression of understory by Oklahoma oak forests. Bulletin of the Torrey Botanical Club 99(6): 293-300.
- Kraus TEC, Dahlgren RA, Zasoski RJ (2003) Tannins in nutrient dynamics of forest ecosystems - a review. Plant and Soil 256(1): 41-66.
- Li Y, Feng Y, Chen Y, Tian Y (2015) Soil microbes alleviate allelopathy of invasive plants. Science Bulletin 60(12): 1083-1091.
- (1973) Soil Survey Staff, Soil survey of Erath County, Texas, U.S. Department of Agriculture, Natural Resource Conservation Service, USA.
- (2004) Soil Survey Staff, Soil survey laboratory methods manual. Version 4.0, U.S. Department of Agriculture, Natural Resource Conservation Service, USA.
- Gee GW, Bauder JW (1986) Particle-size analysis. In Methods of soil analysis, part 1: physical and mineralogical methods, Madison, Wisconsin, pp. 383-411.
- Horvath B, Opara-Nadi O, Beese F (2005) A simple method for measuring the carbonate content of soils. Soil Science Society of America Journal 69: 1066-1068.
- Bremner JM (1996) Nitrogen - Total. In Methods of soil analysis, part 3: chemical methods, Madison, Wisconsin, American Society of Agronomy, Soil Science Society of America, pp. 1086-1121.
- Wang Y, Qian P (2009) Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS One 4(10).
- Klindworth A, Prusse E, Schweer T, Peplies J, Quast C, et al. (2012) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Research 41(1): 1-11.
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7(5): 335-336.?
- Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26(19): 2460-2461.
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, et al. (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72(7): 50695072.
- Koljalg U, Abarenkov NRHK, Tedersoo L, Taylor AFS, Bahram M, et al. (2013) Towards a unified paradigm for sequence-based identification of Fungi. Moleculat Ecology 22(12): 5271-5277.
- Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71: 82288235.
- Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecology 26(1): 32-46.
- Anderson MJ, Crist TO, Chase JM, Vellend M, Inouye BD, et al. (2011) Navigating the multiple meanings of beta diversity: a roadmap for the practicing ecologist. Ecology Letters 14(1): 19-28.
- Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, et al. (2018) Vegan: Community Ecology Package. R package version 2: 4-6.
- R Core Team (2015) R: A language and environment for statistical computing, Vienna, Austria: R Foundation for Statistical Computing.
- Jenny H (1994) Factors of Soil Formation: A System of Quantitative Pedology. Dover Publications Inc, New York, USA.
- Willems A, De Ley J, Gillis M, Kersters K (1991) Comamonadaceae, a new family encompassing the acidovorans rRNA complex, including Variovorax paradoxus gen. nov., comb. nov. for Alcaligenes paradoxus (Davis 1969). International Journal of Systematic Bacteriology 41(3): 445-450.
- Ahn Y (2006) Sustainable nitrogen elimination biotechnologies: a review. Process Biochemistry 41(8): 1709-1721.
- Zhalnina KV, Dias R, Leonard MT, de Quadros PD, Camargo FAO, et al. (2014) Genome sequence of Candidatus Nitrososphaera evergladensis from group I.1b enriched from everglades soil reveals novel genomic features of the ammonia-oxidizing archaea. PloS One 7: 9.
- Tesar M, Reichenauer TG, Sessitsch A (2002) Bacterial rhizosphere populations of black poplar and herbal plants to be used for phytoremediation of diesel fuel. Soil Biology and Biochemistry 34(12): 1883-1892.
- Padmavathi T, Dikshit R, Seshagiri S (2015) Effect of Rhizophagus spp. and plant growth-promoting Acinetobacter junii on Solanum lycopersicum and Capsicum annuum. Brazilian Journal of Botany 38(2): 273-278.
- Anda CCO, Opfergelt S, Declerck S (2016) Silicon acquisition by bananas (c.V. Grande Naine) is increased in presence of the arbuscular mycorrhizal fungus Rhizophagus irregularis MUCL 41833. Plant and Soil 409(1-2): 77-85.
- Merlos MA, Zitka O, Vohtech A, Azcon-Aguilar C, Ferrol N (2016) The arbuscular mycorrhizal fungus Rhizophagus irregularis differentially regulates the copper response of two maize cultivars differing in copper tolerance. Plant Science 253: 68-76.
- Balser TC, Firestone MK (2005) Linking microbial community composition and soil processes in a California annual grassland and mixed-conifer forest. Biogeochemistry 73(2): 395-415.
- Brady JA, Faske JB, Castaneda-Gill JM, King JL, Mitchell FL (2011) High- throughput DNA isolation method for detection of Xyella fastidosa in plant and insect samples. Journal od Microbiology Methods 86(3): 310312.
- Chin KJ, Liesack W, Janssen PH (2001) Opitutus terrae gen nov, sp nov, to accommodate novel strains of the division 'Verrucomicrobia” isolated from rice paddy soil. International Journal of Systemiatic and Evolutionary Microbiology 51(Pt 6): 1965-1968.
- Delwiche CC (1970) The nitrogen cycle. Scientific American 223: 137146.
- Illumina (2013) 16S metagenomic sequencing library preparation.
- Larsen EI, Sly LI, McEwan EG (1999) Manganese (II) adsorption and oxidation by whole cells and a membrane fraction of Pedomicrobium sp. ACM 3067. Archives of Microbiology 171(4): 257-264.
- Nalin R, Normand P, Domenach A (1997) Distribution and N2-fixing activity of Frankia strains in relation to soil depth. Physiologia Plantarum 99(4): 732-738.
- Wang X, Chi C, Nie Y, Tang Y, Tan Y, et al. (2011) Degradation of petroleum hydrocarbons (C6-C40) and crude oil by a novel Dietzia strai. Bioresource Technology 102(17): 7755-7761.
















{kind=link}