




A Validated RP-HPLC Method for Simultaneous Assay Determination and UV-Spectrophotometric Method for Dissolution Estimation in Film-Coated Tablets Containing Cefixime and Clavulanic Acid
Utsav Nepal1,4, Vijay Kumar Panthi2,3,5,6*, Namindra Prasad Chaudhary4,7 and Samip Chaudhary4,8
1Department of Pharmacy, Kathmandu University, Nepal
2Department of Pharmacy, Tribhuvan University, Sunsari Technical College, Nepal
3Research & Development Department, Royal Sasa Nepal pharmaceuticals, Nepal
4Quality Control Department, Royal Sasa Nepal pharmaceuticals, Nepal
5Research & Development Department, Asian Pharmaceuticals, Nepal
6Research & Development Department, Corel Pharmaceuticals, Nepal
7Kantipur College of Medical Science, Tribhuvan University, Nepal
8Tri-Chandra Multiple Campus, Tribhuvan University, Nepal
Submission: July 07, 2023; Published: August 18, 2023
*Corresponding author: Vijay Kumar Panthi, Department of Pharmacy, Tribhuvan University, Sunsari Technical College, Nepal
How to cite this article: Clarence St H. Reframing Vodou’s Veves through Guided Imagery Therapy as Diasporic Expressions Among Haitians in Miami-Dade County, Florida. J Complement Med Alt Healthcare. 2023; 12(1): 555833. DOI: 10.19080/JCMAH.2023.12.555833
Abstract
A simple and novel reverse phase high performance liquid chromatography (RP-HPLC) method was developed for the simultaneous assay estimation of film coated tablets incorporating clavulanic acid (CVA) and cefixime (CFX). Furthermore, the same method was also applied in order to assess the dissolution of CVA while a UV spectroscopic method was produced to examine the drug release of CFX. In this research, an analytical column used was C18 (4.6-mm × 25-cm); 5-μm. Moreover, the mobile phase was prepared by completely dissolving 3.408 gm of disodium hydrogen phosphate in 800 mL of water (HPLC grade), 200 mL methanol was added in (20:80 v/v) it and then finally pH was adjusted to 6.0 using orthophosphoric acid. In addition, flow rate, detecting wavelength, and run time were 1.0 mL/min, 220 nm, and 15 minutes, respectively. Furthermore, the maximum absorption of CFX was obtained at 288 nm thereby this wavelength was optimized for the dissolution estimation of CFX by UV spectroscopy. The validation parameters were validated according to the specifications and guidelines of the international conference on harmonization (ICH) prior to evaluating both assay and dissolution of CVA and CFX. In case of assay estimation, the findings of the linearity revealed a linear interrelation of concentration between 0.16 - 0.25 mg/mL for both CVA and CFX. Similarly, the calibration curve was linear between the concentration range of 0.03 – 0.045 mg/mL and 0.004 – 0.006 mg/mL, respectively for dissolution determination. Additionally, the value of LOD and LOQ of CVA for assay evaluation was obtained 0.011 and 0.033 μg, respectively. The accuracy of the developed method was assessed by determining recovery studies and the mean recovery of CVA and CFX obtained 99.35% and 100.50%, respectively at 100% spiked level for assay analysis. The repeatability evaluation for assay and dissolution demonstrated that the optimized method is precise within the appropriate range and the RSD% was observed lower than 2.0%. In addition, results of different validation parameters including study of solution stability were achieved within the suitable limit as well.
Keywords: Clavulanic acid; Cefixime; Assay; Dissolution; Method validation
Introduction
Cephalosporins demonstrate a broad range of antibacterial activity against Gram positive and Gram-negative bacteria. It possesses the mechanism of action by impeding the synthesis of bacterial cell wall. Cefixime (CFX) trihydrate belongs to the third generation of cephaslosporins which is orally active [1]. Till date, CFX is commercially available in different range of tablets (100, 200, and 400 mg) and suspension. It is administered via oral route for the cure of a variety of infections such as pharyngitis, otitis media, gornorrhoea, infections of lower respiratory tract particularly bronchitis and infections of urinary tract. However, when it delivered at higher doses, patients suffer from different physiological issues including gastrointestinal disorders, headaches, dizziness, and rashes [2]. Thus, from an analytical point of view, it is vital for achieving proper therapeutic concentration in addition to assurance in quality of pharmaceutical formulations [3].
On the other hand, Clavulanic acid (CVA) is beta-lactam molecule having a narrow spectrum of antibacterial activity along with potentiality of irreversible binding with a betalactamases associated with wide range of spectrum [4]. When combinational administration of CVA and other beta-lactamase susceptible antibiotics including ampicillin and amoxicillin was given, the binding characteristics of CVA accelerated the efficacy of the antibiotic against beta-lactamase producing bacteria. Furthermore, in case of analytical determination, previous study has suggested that CVA was quantified by using micro disk agar diffusion technique applying an overnight seed culture of penicillin producing Klebsiella pneumonia [5].
In addition, various research studies have been reported for the simultaneous assessment of CVA and amoxicillin by ultraviolet (UV) spectrophotometry in bulk, and pharmaceutical formulations. Moreover, in previous years, high performance thin layer chromatography (HPLC) method was suggested for the simultaneous evaluation of CVA and CFX in synthetic mixture [6]. Additionally, a validated stability indicating HPLC method was also suggested for the concurrent determination of amoxicillin and CVA in injection [7]. However, it is still very difficult to find an appropriate HPLC method for the simultaneous and separate analysis of CVA and CFX. Similarly, at present, no HPLC method is reported for the simultaneous estimation of CVA and CFX in a tablet dosage form. Therefore, in this research, a simple, robust, sensitive, precise, and suitable stability indicating method is suggested for simultaneous assay determination by HPLC and separate dissolution analysis of CVA and CFX by HPLC and UV, respectively.
Materials and Methods
Chemicals and Reagents
A film coated tablet incorporating cefixime and potassium clavulanate was received from the research and development department of our pharmaceutical company. The HPLC grade of both methanol and water, and the material of analytical reagent grade including disodium hydrogen phosphate, and potassium dihydrogen were purchased from Thermo Fisher Scientific India Ltd.
Instrumentation
The below mentioned instruments were used in order to analyze the formulated film coated tablets. The HPLC (Agilent 1260, Waldron, Germany) system having a pump (model; G7116A), model of autosampler (G7129A) and the column dimension was COSMOSIL C18 (250 cm × 4.6 mm) 5 μm (Paisley, UK) applied for the method development of simultaneous assay determination for CVA and CFX and dissolution determination of CVA. Moreover, the detector used was UV/VIS which operated at 220 nm. In addition, the used software was Agilent OpenLab (Version 3.2.0.0) in order to process and evaluate the data. Furthermore, for weighing of reagents, analytical balance revealing four digits on screen was used (Radwag, model: AS 220.X2, Poland) and sonicator (MRC, model: ACP-150H, India) was applied to dissolve the reagents. Similarly, for dissolution determination of CFX, UV-VIS spectrophotometer (Agilent Cary 60) was used.
Analytical method validation for simultaneous assay determination of CFX and CVA
Chromatographic conditions
For preparation of diluents, 7.10 gm of disodium hydrogen phosphate was completely dissolved in 500 mL water with continuous stirring and the pH was adjusted to 7.0 with adding solution of potassium dihydrogen phosphate. Moreover, the preparation of the mobile phase was done by adding 3.408 gm of disodium hydrogen phosphate in water (HPLC grade) containing 800 mL and dissolved completely. In addition, 200 mL methanol was added to it and then final pH was adjusted to 6.0 with orthophosphoric acid. After completion of mobile phase preparation, it was filtered using 0.45 μm membrane filters and degassing was performed by sonication for 20 min. The testing was carried out on an Agilent 1260 HPLC system and the analytes were performed on an analytical column C18, (5 μm, 250 x 4.6 mm). The detection wavelength and the detector were 220 nm and UV/VIS, respectively. Additionally, the column temperature was operated at 30oC. The volume for injection was 1.0 mL/min while flow rate was 20 μL and the run time was 15 minutes.
Preparation of Standard Solution
Accurately weighed about 20 mg reference standard of each CFX trihydrate and CVA then transferred into 100 mL volumetric flask. Subsequently, 60 mL diluent was added and sonicated for 20 minutes. Finally, volume was making up to the mark; the solution was properly shaken and filtered.
Preparation of Test Solution
The individual weight of 20 tablets was taken and crushed to obtain a powder. The quantity of crushed powder equivalent to 0.4 g of CFX was transferred to 100 mL volumetric flask then diluent was added, sonicated and finally volume was made up with the same solvent. Additionally, pipette out 5 mL of this solution and further volume make up was done to 100 mL with diluent, properly shaken and filtered.
Analytical method validation for dissolution estimation
Chromatographic conditions for CVA
The dissolution estimation of CVA tablets was carried out by using USP apparatus 2 (paddle). The drug release profile of this drug was performed in dissolution media (purified water). After the temperature reached 37 ± 0.5oC, a single tablet was added individually to separate dissolution dishes containing 900 mL of dissolution medium and run for 30 minutes at 75 rpm. Subsequently, 5 mL of the solution was withdrawn using syringes from each dissolution container at predetermined time intervals of 15, 30, 45, and 60 minutes. After each withdrawal, 5 mL of fresh dissolution media was added to maintain sink condition. The chromatographic conditions used for the dissolution assessment of CVA were mentioned in section 2.3.1. Moreover, for preparation of test solution, pipette out suitable volume of the dissolution medium from each vessel and filter through 0.2 μm membrane filter. Then 5 mL of the filtered solution was further diluted to 10 mL with dissolution medium. Furthermore, in order to prepare a standard solution, accurately the 15 mg reference standard was taken and diluted with a 200 mL dissolution medium. Finally, 5 mL of this standard solution was taken and further diluted with 10 mL dissolution medium, filtered then measured the chromatogram of both solutions. The average results acquired from triplicate quantifications were used in order to calculate the percentage of dissolved CVA and CFX by applying the below mentioned formula [8,9] % of dissolved drug Formula (1).
Spectroscopic conditions for CFX
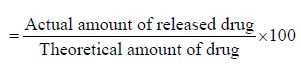
The dissolution estimation of CFX tablets was carried out by using USP apparatus 1 (basket). The drug release profile of CFX was carried out in dissolution media containing 0.05M potassium phosphate buffer (pH 7.2). In addition, the volume of dissolution medium, basket speed and run time were 900 mL, 100 rpm, and 45 minutes, respectively. After completion of run time, suitable volume from the dissolution vessel was taken and filtered. Additionally, 5 mL of the filtrate was further diluted to 100 mL with dissolution medium as a test solution. The maximum absorbance was measured at about 288 nm then content of CFX in the medium was calculated from the absorbance obtained from the standard solution of CFX having a concentration of 0.005 mg/mL which was prepared by dissolving about 20 mg of reference standard of CFX in 200 mL of dissolution medium. Pipette out 5 mL of this solution then further diluted to 100 mL.
Parameters of Analytical method validation for Assay and Dissolution estimation
Specificity
This is one of the important parameters of analytical method validation which ensures that there is no interference between the analyte and other components in a formulation or complex mixture [10]. In this study, for determination of assay, specificity was evaluated by separately injecting one blank solution, one placebo solution, and five replicates of each standard and test sample of both CFX and CVA at 100% concentration. The retention time of standard samples of CFX and CVA were identified and compared with their test sample. Moreover, in case of dissolution study, specificity was evaluated by separately injecting one placebo solution, and five replicates of each standard and test sample of both CVA at 100% concentration. The retention time of standard samples of CVA were identified and compared with their test sample. Similarly, in case of CFX, the absorbance of standard samples was noted and compared with their test one.
Linearity and Range
Linearity is defined as the potentiality to receive test results that have concentration dependent relationship of the analyte [11,12]. This parameter was examined by three injections of five different concentrations of each CFX and CVA. In this research, in case of assay determination, the concentrations of each CFX and CVA were 0.16, 0.18, 020, 0.22, and 0.25 μg/ml. The mean value of peak areas was represented against concentrations of both CFX and CVA. Finally, linearity is assessed by applying the calibration curve in order to find out the coefficient of correlation, slope, and intercept. Similarly, the linearity study was carried out for examining the CVA sample dissolution from five different concentrations (0.03, 0.034, 0.038, 0.041, 0.045 μg/mL) and the recovery % of each concentration was reported then this parameter was evaluated by visual inspection of a plot area as a function of concentration. Finally, the correlation coefficient was calculated. Similarly, in order to assess the linearity study of CFX, five different concentrations of CFX (0.004, 0.0045, 0.005, 0.0055, 0.006 μg/mL) was determined then the recovery % of each concentration was evaluated by calibration curve of absorbance and finally the correlation coefficient was examined.
Accuracy
This parameter deals with the closeness of the predicted value and the obtained results. In this study, the accuracy for the assay method was examined by recovery studies at three different concentration levels in which the corresponding concentration levels were 80%, 100%, and 120%. The three measurements were injected for each concentration level of CVA and CFX. The recovery % of both drugs was added and the value of RSD was estimated for each of the injected samples. In this study, accuracy of the dissolution method was assessed by recovery studies at concentration levels of 80%, 100%, and s120% of both CVA and CFX. The replicates of both CVA and CFX were evaluated separately with respect to area and absorbance, respectively. In addition, percentage recovery of both CFX and CVA were added, and RSD were calculated for each of the replicate samples.
Precision
In this study, system precision for assay determination of both CVA and CFX were evaluated by injecting five standard solutions at the 100% concentration levels on the same day. Furthermore, method precision was examined with replicates of six assay measurements of the sample solution at the 100% concentration levels on the same day then the RSD% of the obtained results were calculated in order to determine the repeatability values. Similarly, in this research, system precision and method precision (repeatability) of both CVA and CFX for dissolution estimation was evaluated with five replicates of standard and six replicates of dissolution sample at 100% concentration in accordance with area and absorbance, respectively. Moreover, method precision was also determined by varying analysts and days at the 100% concentration level. The RSD% of the obtained results was calculated prior to assessing the repeatability findings.
Determination of LOD and LOQ
The sensitivity of the assay and dissolution method was evaluated by repeating calibration curve for 6 times and SD of the intercepts was examined by applying below mentioned formula in order to find out the result of LOD and LOQ.
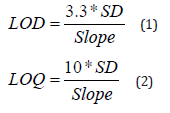
Where, SD = standard deviation of Y- intercept of 6 calibration curves.
Slope = mean slope of the 6 calibration curves [13].
Robustness
As per the definition given by ICH, this parameter deals with the capability of analytical methods to be not influenced by minor modifications [10]. In this research, robustness for assay evaluation of both CVA and CFX were assessed by allowing minor variations in flow rate and pH of mobile phase. The flow rate was varied by ± 0.4 mL/min while pH was examined by applying ± 0.4. The value of assay and %RSD of each sample were determined. Moreover, the robustness of CVA for the dissolution method was evaluated by allowing minor alterations in the chromatographic conditions. The applied conditions were flow rates and pH of mobile phase which varied by ± 0.2 mL/min and ± 0.2, respectively. The recovery % and RSD% of each sample were analyzed. Accordingly, for estimation of CFX dissolution, the robustness was studied with different detection wavelengths. The detection wavelength was altered by ± 2.0 nm then the recovery% and RSD% were evaluated.
Solution Stability
The assay stability of both CVA and CFX was evaluated by assessing the standard and test samples at 0 h. In addition, both standard and test samples were also kept in the refrigerator and at room temperature (30°C) for 24 h. The three replicates from individual solutions were evaluated and the mean value of peak and the RSD were calculated.
Statistical Analyses
Statistical analyses were done using GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA). The obtained dissolution data were assessed with one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test and expressed as mean ± standard deviation. In comparative dissolution analysis, p < 0.05 was applied to indicate statistical significance.
Results and Discussion
Analytical method development and optimization
The physical and chemical properties of both CVA and CFX were studied from previously published resources. In this study, analytical column C18, (5 μm, 250 x 4.6 mm) was used for the separation of chromatograms in order to evaluate simultaneous assay of CVA and CFX in addition with dissolution estimation of CVA. In contrast, UV spectroscopy was used for dissolution determination of CFX. In this study, a solution consisting properly dissolved 3.408 gm of disodium hydrogen phosphate in 800 mL water (HPLC grade) in which 200 mL methanol was added (in the proportion of 80:20 v/v) and final pH was adjusted to 6.0 with orthophosphoric acid is used as mobile phase. The detection wavelength and the detector were 220 nm and UV/VIS, respectively. Moreover, the column temperature was operating at 30ºC. The volume for injection was 20 μL, 1.0 mL/min flow rate and run time applied 15 minutes was optimized as the suitable method for the simultaneous assay estimation of film coated tablets containing CVA and CFX. In addition, this method was also applied for the dissolution assessment of CVA whereas a UV spectrophotometer having detection wavelength 288 nm was used for the dissolution estimation of CFX. The maximum absorbance was observed in 288 nm of UV spectroscopy thereby it is optimized as the final wavelength for dissolution determination of CFX (Figure 5).
Specificity
In this study, in order to evaluate the specificity of the assay method, the standard and test solution of each CVA and CFX, blank, and placebo solution were separately injected at 100% concentration. The total five measurements of both standard and sample solution of each CVA and CFX were injected, and the injected volume of all above solutions was 20 μL. The chromatograms that are obtained after each sample evaluation were shown in Figure 1, 2, 3, and 4. The HPLC peak demonstrated that no close peak was detected to the retention time of both standard and test solution for CFX and CVA interference, which revealed the specificity of the method. Additionally, retention time data of standard and sample solution was demonstrated in Table 2 and 3.







Furthermore, procedures for specificity determination of dissolution methods of both CVA and CFX were mentioned in section 2.5.1. The HPLC peak demonstrated that there were no coeluting peaks detected to the retention time of standard and sample solution of CVA. Furthermore, the retention time was compiled for the major peak of each standard and test sample of CVA which was presented as a figure in Supplementary File 1 and 2. Additionally, the retention time and the area of both standard and sample solutions of CVA are mentioned in Table 4. Thus, these observations show the specificity of the dissolution method for CVA evaluation. Moreover, in case of CFX, the absorbance value of standard and sample solution is very close to each other which confirms the validated specificity of the method (Figure 5). Furthermore, findings of standard and sample absorbance for dissolution determination of CFX were demonstrated in Supplementary File 3.




Linearity
In case of verification of analytical procedure, the term linearity means the potentiality of the analytical method to get test data which have linear relationship with concentration of, between the range. In this study, linearity of both CVA and CFX assay method is determined by taking mean peak area from HPLC and represented with respect to the concentrations prior to achieving the calibration curve. In case of assay estimation, the findings of the linearity revealed a linear interrelation of concentration between 0.16 - 0.25mg/mL for both CVA and CFX (Figure 6 and 7). In addition, the correlation coefficient and regression equation of CVA were 0.993 and y = 46587x - 755.2, respectively. Accordingly, in CFX determination, values of R2 and regression equation were 0.995 and y = 46587x-755.2, respectively, showing a direct association between the analyte concentration and their peak area.


Additionally, in case of dissolution, the linearity of CVA and CFX is examined by HPLC and UV spectroscopy. The detailed procedure is mentioned section in 2.5.2. The correlation coefficient and regression equation of CVA were 0.998 and y = 34203x, respectively (Figure 8) Moreover, R2 value and regression equation of CFX were 1.00 and y = 60.42x, respectively (Figure 9).
LOD and LOQ
The sensitivity of the analytical method was evaluated by assessing LOD and LOQ as per the formula illustrated in section 2.5.5. In this study, the LOD and LOQ of CVA in assay determination were found to be 0.011 and 0.033 μg, respectively. Similarly, LOD and LOQ of CFX in assay evaluation were 0.0086 and 0.026 μg, respectively. Moreover, in case of dissolution estimation, the LOD and LOQ of CVA were 0.0008 and 0.0025μg, respectively. Furthermore, LOD and LOQ of CFX in dissolution determination were 0.00001 and 0.00003μg, respectively.


AccuracyIn this research, two precision including system and method (repeatability and intermediate) were assessed. In case of determination of system precision for assay analysis, all the evaluated parameters were within the limit. The RSD% and number of theoretical plates were NMT 2.0% and NLT 1000, respectively. The obtained results were demonstrated in Table 9 and 10. Similarly, the system precision for dissolution assessment of CVA was presented in Table 11. In addition, in terms of evaluation of method precision, the observed assay value of RSD% for CVA and CFX in analysis of method precision was all achieved lower than 2.0%, as mentioned in Table 12. Accordingly, determination of method precision for dissolution analysis of CFX was shown in Supplementary File 4. In addition, the obtained RSD values less than 2.0% indicate the precision of the method [14]. Therefore, the findings obtained from system and method precision has revealed that the validated method is within the suitable range.










The accuracy in the analytical method is defined as the vicinity of the obtained value by that method as compared with the true value [13]. The overall method of accuracy for assay and dissolution is represented in section 2.5.3. In this research, the accuracy study exhibited percentage recovery at all three spiked level (80%, 100%, and 120%) between 99.33% -101.58% and the value of %RSD were in the range of 0.032 – 0.648 in assay determination for both CVA and CFX (Table 5 and 6).
Moreover, in the case of dissolution assessment, accuracy study also revealed percentage recovery in the range of 98.55 – 101.20%. Similarly, in this parameter the obtained value of % RSD was 0.085% - 0.426% at all three spiked levels. The overall findings are demonstrated in Table 7 and 8. The value of percentage recovery and %RSD were achieved within the acceptable range (98.0% to 102.0%). Similarly, the results of %RSD were obtained within the limit (NMT 2.0%). These observed results showed the accuracy of the method for assay and dissolution estimation of CVA and CFX.
Precision
In this research, two precision including system and method (repeatability and intermediate) were assessed. In case of determination of system precision for assay analysis, all the evaluated parameters were within the limit. The RSD% and number of theoretical plates were NMT 2.0% and NLT 1000, respectively. The obtained results were demonstrated in Table 9 and 10. Similarly, the system precision for dissolution assessment of CVA was presented in Table 11. In addition, in terms of evaluation of method precision, the observed assay value of RSD% for CVA and CFX in analysis of method precision was all achieved lower than 2.0%, as mentioned in Table 12. Accordingly, determination of method precision for dissolution analysis of CFX was shown in Supplementary File 4. In addition, the obtained RSD values less than 2.0% indicate the precision of the method [14]. Therefore, the findings obtained from system and method precision has revealed that the validated method is within the suitable range.
Robustness
This parameter was performed by evaluating the effect of minor alternations in HPLC and UV spectroscopy. The findings of this parameter revealed slight changes in analyzing conditions in these two instruments. The modifications done in HPLC conditions were flow rate and pH of mobile phase while wavelength was slightly altered in case of UC spectroscopy. In assay evaluation of CVA and CFX, the observed RSD% was within suitable range (NMT 2.0%). At 0.8 mL/min flow rate, the assay of CVA and CFX were 98.63 and 98.61, respectively. Furthermore, at pH 6.2, the %RSD of CVA and CFX were 0.1485, and 0.4398, respectively. The obtained results of assay determination with robustness study were observed within the limit which is demonstrated in Table 13 and 14.
Accordingly, in case of dissolution determination, the RSD% of CVA at 1.2 mL/min flow rate and at pH 6.2 was 0.4688 and 0.1683, respectively. In addition, the dissolution% of CFX in 288 and 292 wavelengths was 101.29 and 100.22, respectively. The obtained results were revealed in detail in Table 15 and 16. Despite minor changes in flow rate, pH of mobile phase, and wavelength there were no notable findings which demonstrated the method is robust for dissolution determination.
Solution Stability
In this study, both CVA and CFX showed good stability for 24 hr at 30°C and 4°C. During initial analysis, the CVA revealed 101.02% assay while it demonstrated 100.61% and 100.76% assay after 24 hr at 30°C and 4°C, respectively. Similarly, CFX showed 100.77% assay during initial analysis whereas it exhibited 99.38% and 100.90% assay after 24 hr at 30°C and 4°C, respectively. Moreover, in solution stability analysis, tailing factors and the number of theoretical plates were also obtained within the appropriate limits. In addition, the percentage recovery was observed within the acceptable limits (98.0% to 102.0%) and the RSD% was also obtained below 2.0%, indicating a better stability of products in two different storage storage conditions. The observed findings were exhibited in Table 17 and 18.






Assessment of Dissolution
In this research, the dissolution of our formulated sample containing CVA and CFX was compared with the innovator brand. The method of determination was mentioned in section 2.4.1. At 60 minutes, CVA showed 97.41% of drug release in purified water whereas the drug release of the market sample was 92.22%. Moreover, at 45 minutes, our formulated sample exhibited 102.25% dissolution of CFX in dissolution media containing 0.05M potassium phosphate buffer (pH 7.2) while the market sample showed 99.27% drug release in the same dissolution medium. The dissolution profile of both drugs in comparison with the market was demonstrated in Figure 10. In addition, in this study, the dissolution value of our sample was statistically insignificant (P > 0.05) in different time periods in comparison to the market sample.

Conclusion
In this research, a rapid, sensitive, and simple HPLC and UV method has been developed with better accuracy and precision. In addition, there is no requirement of any laborious and timeconsuming measurement and preparation technique. This method can be readily applied for the routine drug estimation of filmcoated tablets containing both CVA and CFX. Moreover, the notable findings of the produced method are shorter run and retention time. The RP-HPLC method can be applied for the simultaneous determination of assay of both CAV and CFX. Accordingly, the same method was also employed in order to examine the dissolution of CVA, and a simple UV method has been developed for the dissolution estimation of CFX. All the validation parameters were validated according to the guidelines of the international conference on harmonization. Moreover, this validated analytical method can serve as a facilitator to the pharmaceutical companies and other research institutions to assess various pharmaceutical dosage forms incorporating CVA and CFX in order to examine their quality in products.
Acknowledgements
All authors are grateful to Royal Sasa Nepal Pharmaceuticals, Chitwan, Nepal for facilitating opportunity to carry out this research.
References
- Brogden RN Campoli Richards DM (1989) "Cefixime. A review of its antibacterial activity. Pharmacokinetic properties and therapeutic potential" Drugs 38(4): 524- 550.
- Azmi SNH, Iqbal B, Al-Humaimi NSH, IAl-Salmani IRS, Al-Ghafri NAS (2013) "Quantitative analysis of cefixime via complexation with palladium (II) in pharmaceutical formulations by spectrophotometry," J Pharm Anal 3(4): 248- 256.
- Meng F, Chen X, Zeng Y, Zhong D (2005) Sensitive liquid chromatography-tandem mass spectrometry method for the determination of cefixime in human plasma: application to a pharmacokinetic study," Journal of chromatography. B, Analytical technologies in the biomedical and life sciences 819(2): 277-282.
- Nelson JD, Kusmiesz H, Shelton S (1982) "Pharmacokinetics of potassium clavulanate in combination with amoxicillin in pediatric patients," Antimicrob Agents Chemother 21(4): 681-682.
- Hunter PA, Coleman K, Fisher J, Taylor D (1980) "In vitro synergistic properties of clavulanic acid, with ampicillin, amoxycillin and ticarcillin," The Journal of antimicrobial chemotherapy 6(4): 455-470
- Khan IU, Sharif S, Ashfaq M, Asghar MN (2019) "Simultaneous Determination of Potassium Clavulanate and Cefixime in Synthetic Mixtures by High-Performance Liquid Chromatography," Journal of AOAC International 91(4): 744-749.
- Mallikarjuna D, Tippa R, Singh N (2010) "Development and Validation of Stability Indicating HPLC Method for Simultaneous Estimation of Amoxicillin and Clavulanic Acid in Injection," American Journal of Analytical Chemistry 01.
- Rivera-Leyva JC, García-Flores M, Valladares-Méndez A, Orozco-Castellanos LM, Martínez-Alfaro M (2012) "Comparative Studies on the Dissolution Profiles of Oral Ibuprofen Suspension and Commercial Tablets using Biopharmaceutical Classification System Criteria," Indian journal of pharmaceutical sciences 74(4): 312- 318.
- Panthi VK, Jha SK, Chaubey R, Pangeni R (2021) "Formulation and development of Serratiopeptidases enteric coated tablets and analytical method validation by UV Spectroscopy," Int J Anal Che 9749474.
- Alquadeib BT (2019)"Development and validation of a new HPLC analytical method for the determination of diclofenac in tablets," Saudi Pharmaceutical Journal 27(1): 66- 70.
- Naseef H, Moqadi R, Qurt M (2018)"Development and Validation of an HPLC Method for Determination of Antidiabetic Drug Alogliptin Benzoate in Bulk and Tablets," Journal of Analytical Methods in Chemistry 1902510.
- Nepal U, Panthi VK, Chaudhary NP, Chaudhary S (2022) "A Validated RP-HPLC Method for Simultaneous Determination of Cefixime and Clavulanic Acid Powder in Pediatric Oral Suspension," Int J Anal Chem 8331762.
- Panthi VK, Nepal U (2022) "Formulation and Development of a Water-in-Oil Emulsion-Based Luliconazole Cream: In Vitro Characterization and Analytical Method Validation by RP-HPLC," Int J Anal Chem 7273840.
- Maleque M, Hasan MR, Hossen F, Safi S (2012) "Development and validation of a simple UV spectrophotometric method for the determination of levofloxacin both in bulk and marketed dosage formulations," J Pharm Anal 2(6): 454- 457.















