




Clinical and Laboratory Study on Children with Glycogen Storage Disease Type-1 in Upper Egypt
Tahia H Saleem1, Hamdy N Eltalawy2, Nagla H Abu-faddan3*, Ahmed E Ahmed4, Yasser Gamal5 and Mohammed H Hassan6
1Biochemistry Department, Assuit Universty, Egypt
2Neuropsychiatry Department, Assuit Universty, Egypt
3Pediatric Department, Assuit Universty, Egypt
4Pediatric Department, Qena Universty, Egypt
5Pathology Department, Assuit Universty, Egypt
6Biochemistry Department, Qena Universty, Egypt
Submission: September 14, 2016; Published: September 28, 2016
*Corresponding author: Nagla Abu Faddan, Assiut University, Assiut University Children Hospital, Egypt.
How to cite this article: Tahia H S, Hamdy N E, Nagla H A-f, Ahmed E A, Yasser G, et al. Clinical and Laboratory Study on Children with Glycogen Storage Disease Type-1 in Upper Egypt. Adv Res Gastroentero Hepatol. 2016; 2(1): 555578. DOI: 10.19080/ARGH.2016.02.555578
Abstract
Background: One of the GSDs that principally affect the liver is Type 1 GSD. GSDI patients may present with fast-induced hypoglycemia and hyperlactacidemia. More commonly, the first symptom is the presence of a protruded abdomen due to marked hepatomegaly.
Study aims: This study aims to assess frequency, clinical manifestations, biochemical features of GSD1 in children attending University Hospitals in Upper Egypt, and to identify the possible role of plasma biotinidase in diagnosis of GSD-1 in these children.
Patients and Methods: Any child with unexplained hepatomegaly with growth retardation, early fasting hypoglycemia, hyperuricemia, or hyperlipidemia during the period from July 1st 2011 up to December 31st 2013 was included in this study. Complete blood counts, liver function tests, Prothrombin time and concentration, serum glucose, cholesterol, triacylglycerol, plasma biotinidase, Plasma lactate and plasma uric acid level were measured. Liver biopsy was done to patients when needed and Glucose-6-phosphatase activity was measured in biopsied liver tissue.
Results: GSD-1 could be detected in only 10 children. 50% of them males and 50% females, with the mean age 4.8+4 SD years, 8(70%) between 1 to 6 years old. The histopathological diagnosis of GSD-I was confirmed. There was lower glucose-6-phosphatase activity in the biopsied liver tissue homogenates and high plasma biotinidase was detected in these 10 children.
Conclusion: GSD-1 should be considered in all children with unexplained hepatomegaly. Elevated serum biotinidase can be used as a diagnostic marker for hepatic glycogen storage disorders and Biotin supplementation in children with GSD-1 is very important.
Keywords: Type 1 GSD; Children
Abbreviations: PPV: Positive Predictive Value; NPV: Negative Predictive Value; GSD: Glycogen Storage Disease; SPSS: Statistical Science for Social Package
Introduction
Because liver has central role in synthetic, degradative and regulatory pathways involving carbohydrate, protein, lipid, trace elements and vitamin metabolism many metabolic abnormalities affect the liver [1]. One of the inherited metabolic disorders that principally affect the liver is Glycogen storage disease Type 1 (GSD-1), which is caused by the absence or deficiency of glucose- 6- phosphatase activity in the liver this leads to inadequate conversion of the glucose - 6- phosphate to glucose and makes affected individuals susceptible to fasting hypoglycemia [2]. GSD-I patients may present with fast-induced hypoglycemia (sometimes occurring rapidly in about 2 to 2 and a half hours after a meal) and hyperlactacidemia in the neonatal period. More commonly, the first symptom is the presence of a protruded abdomen due to marked hepatomegaly around 3 months of age, though in some cases the liver may already be enlarged at birth.
Fasting tolerance is very limited: hypoglycemia, which may cause convulsions, and lactic academia, account for the initial gravity of the disease [3,4]. Early diagnosis and treatment is important for improving quality of life, reducing the damaging effects on organs that become engorged with glycogen, and extending the patient’s life span [5]. Clinical and laboratory evidence often guide the evaluation. Liver biopsy offers morphologic study and permits enzyme assay. Genetic diagnostic approach also available [1]. Biotinidase, the biotin recycling enzyme is ubiquitously distributed and occurs at high levels in the liver, serum, and kidney. It is synthesized by the liver and secreted into the blood [6]. An increase in glucose-6-P or lactate act as activators for biotinidase and this is the cause of the observed increase of biotinidase in plasma of patients with GSD-1. This suggests the possibility of diagnosis or at least strong support for the diagnosis of GSD-1 and liver biopsy may be avoided [6,7]. Aim of the work: This study aims to assess frequency, clinical manifestations, biochemical features of GSD-1 in children attending Assiut University children Hospital, pediatric departments of Sohag and Qena university hospitals, Upper Egypt and to identify the possible role of plasma biotinidase as a rapid non invasive and conventional biochemical marker in the blood for diagnosis of GSD-1 in these children.
Patients and Methods
This cross sectional, hospital based study was cARGHied out from July 1st 2011 up to December 31st 2013 in Assiut University children Hospital, pediatric departments of Sohag and Qena university hospitals, Upper Egypt. This study was approved by the Ethical Committee of Faculty of Medicine, Assiut University, according to the latest revision of Declaration of Helsinki and informed consent was obtained from participant’s parent/legal guardian. The inclusion criteria of this study were any child with hepatomegaly with clinical suspicion of having a metabolic disorder e.g. growth retardation, failure to thrive, dimorphic features or seizures. Children with proven infectious, hematologic, toxic, autoimmune or even malignant cause of hepatomegaly were excluded from this study. During the study period all children with the inclusion criteria were included in this study, they were 40 children (25 male and 15 female). Full clinical assessment was performed to all patients including thorough history and clinical examination. All children included in this study were subjected to the following: abdominal ultrasonography, complete blood counts, liver function tests, Prothrombin time and concentration, serum glucose, plasma cholesterol, plasma triacylglycerol, plasma lactate, plasma uric acid level. Plasma biotinidase level was measured by using enzyme-linked immune-sorbent assay (ELISA) multiskan EX micro plate photometer, thermo scientific, STAT FAX-2100, USA(Glory Science Co., Ltd, CATALOG #: 95562, USA). Under sonographic guided percutaneous needle liver biopsy were done to patients when needed: Variable liver tissue cores ranging from 0.3 to 2 cm in length were taken from the pediatric patients using spring loaded biopsy needle “18G Х 16 cm” GTA, LP 0019\12-Italy. Part of the biopsy core was embedded in 10% formalin for histopathological examination using Hematoxylin & Eosin “H&E” stain. Diagnosis of glycogen storage disease based on the presence of mosaic pattern of the hepatocytes with the presence of periodic acid-Schiff staining “PAS” positivediastase sensitive inclusions which are glycogen deposits in liver biopsies of these children [8]. Glucose-6-phosphatase activity was measured in biopsied liver tissue homogenates according to Koide & Oda [9] and King [10] and total proteins Spectrum Diagnostics total protein reagent “Biuret reagent”(CATALOG #: 310 001, Germany) have been measured in the homogenate using T60 UV visible spectrophotometer. Then the enzyme activity in units was divided by the total protein per mg tissue to give the enzyme activity in units/mg tissue protein.
Statistical Analysis
Statistical Science for Social Package (SPSS V12, SPSS Inc., Chicago, IL, USA) was used for data analysis. Data were presented as mean (SD) or number (%) as appropriate. For comparison of two groups, the parametric ‘Student’s t test’ and non-parametric ‘chi squared test’ for independent variables were used. For all tests, a probability (p) <0.05 was considered significant. To detect if the serum biotinidase can be used as diagnostic marker in children with GSD-1, Sensitivity (ability of the test to detect +ve cases), Specificity (ability of the test to exclude negative cases), positive predictive value (PPV) and negative predictive value (NPV) were calculated.
Results
Among the 40 children included in this study GSD-1 could be detected in only 10 children. They were 5 (50%) males and 5 (50%) females, with the mean age 4.8+4 SD years and age range 0.025-16 years. The histopathological diagnosis of GSD-I was confirmed in all these 10 children. This based on the presence of mosaic pattern of the hepatocytes with the presence of periodic acid-Schiff staining “PAS” positive- diastase sensitive inclusions which are glycogen deposits in liver biopsies of these children [11]. The main histopathological findings of the GSD-I patients involved in this study were in the form of fatty changes, nuclear hyperglycogenation and fibrosis. They were graded 0-3 according to Gogus et al. [11]. All GSD-1 patients showed fatty changes and nuclear hyperglycogenation in varying degrees from grade 1 to grade 3 but as regard the degree of fibrosis, two cases show no fibrosis (grade 0), one case showed massive fibrosis merging to cirrhosis (grade 3) and the remaining cases showed mild to moderate fibrosis in varying degrees from grade 1 to grade 2. There was also, lower glucose-6-phosphatase activity in the biopsied liver tissue homogenates of these 10 children. According to clinical, laboratory and histopathological findings, all children included in this study were divided into two groups:
- Group A: contains 10 pediatric patients with the provisional and final diagnosis of glycogen storage disease GSD- 1.
- Group B: contains 30 children with the provisional diagnosis of glycogen storage disease according to the inclusion criteria but the final diagnosis wasn’t glycogen storage disease.
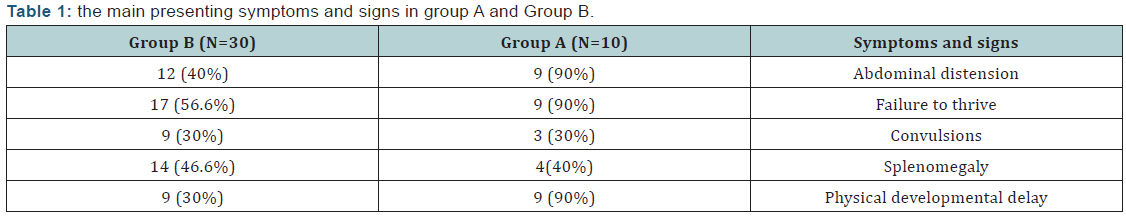
(Table 1) shows the main presenting symptoms and signs in group A and Group B.
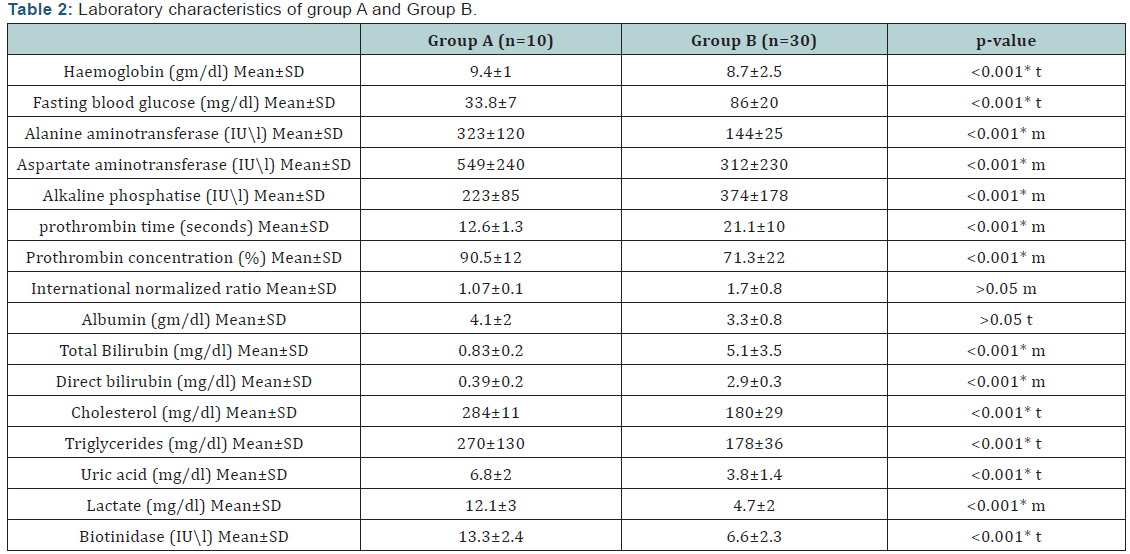
(Table 2) shows Laboratory characteristics of group A and Group B.
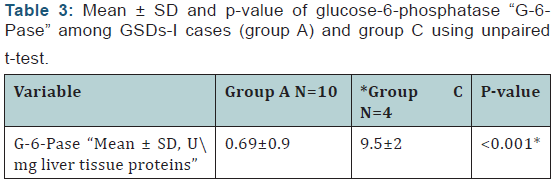
(Table 3) shows comparison between of glucose-6- phosphatase “G-6-Pase” among GSDs-I cases (group A) and control group.
The results of this study show that plasma biotinidase has positive predictive value 80%, negative predictive value 95%, sensitivity 91% and specificity 74% in GSD-I patients.
Discussion
Based on clinical, biochemical and histopathological findings 10 children proved to have GSD-I in this study. They were 5 males & 5 females with no statistically significant difference regarding sex. In agreement with these findings, previous studies [12,13] reported that the sex ratio among the studied GSD-I patients was 1:1. Regarding the clinical presentation in the present study, abdominal distension, failure to thrive and physical developmental delay were present in 90% of GSD-I patients, convulsions were present in 30% of them and hepatomegaly was present in all GSD-I patients (100%). This is in accordance with previous studies [13-16] who reported that the main complaint in GSD-1 patients was abdominal protruding with different rate from 57.8%- 83.3% of cases, followed by growth retardation in 40%- 50% of cases and hepatomegaly was present in 100% of cases. The main laboratory findings of GSD-I patients involved in this study, fasting hypoglycaemia and anemia, have the highest frequency (100%), followed hyperlipidemia (80%) followed by hyperuricemia (70%), while hyperbilirubinemia has the lowest frequency (30%). In agreement with these findings, previous studies [14,16,17] reported that hypoglycemia, hypertriglyceridemia, lactic acidosis, hyperuricemia were the most frequent laboratory findings among GSD-I patients. Also Carvalho et al. [18] found that all patients with GSD-1 in their study had anemia.
Among GSD-I patients in this study, there were statistically significant raised transaminase. These results differ from what reported by Priya et al. [2] who stated that despite marked hepatomegaly, the liver transaminase levels are usually normal or only slightly elevated. But in accordance with the results of other studies [14,16] who found that there was a consistent increase of amino transferases in serum among GSD-1 patients involved in their studies. Glycogen storage leads to hepatocytes injury with subsequent elevation of liver transaminase [19].
In the present study, the plasma biotinidase activity in patients with GSD-I showed statistically significant increase versus the other group. Also the results of this study revealed that plasma biotinidase is considered better positive than negative in prediction of GSD-I with higher sensitivity and low false negative rate. In agreement with these findings, the results of previous studies [15,20,21] who reported that markedly elevated serum biotinidase were found in cases of GSD-1, concluding that GSD-1 should be considered in children with elevated serum biotinidase activity. Also, Paesold Burda et al. [22] proposed from the results of their study that elevated serum biotinidase can be used as a diagnostic marker for hepatic glycogen storage disorders.
Regarding the liver biopsy, the histopathological diagnosis of GSD-1 was confirmed in all 10 children involved in group 1. This based on the presence of mosaic pattern of the hepatocytes with the presence of periodic acid-Schiff staining “PAS” positivediastase sensitive inclusions which are glycogen deposits in liver biopsies of these children8. The main histopathological findings of the GSD-1 patients involved in this study were in the form of fatty changes, nuclear hyperglycogenation and fibrosis. They were graded 0-3 according to Gogus et al. [11]. All GSD-1 patients showed fatty changes and nuclear hyperglycogenation in varying degrees from grade 1 to grade 3 but as regard the degree of fibrosis, two cases show no fibrosis (grade 0), one case showed massive fibrosis merging to cirrhosis (grade 3) and the remaining cases showed mild to moderate fibrosis in varying degrees from grade 1 to grade 2. These findings are in accordance with Saltik et al. [15] who reported that the histopathological findings of the liver included fibrosis (75.6%), steatosis (37.8%), mosaicism (24.4%) and nuclear hyperglycogenation (15.6%) among GSD-I involved in his study. There was also, highly statistically significant lower glucose-6-phosphatase activity when measured in the biopsied liver tissue homogenates of the GSD-1 patients when compared with the control group. This is in accordance of other studies who reported that diagnosis of GSD-1 is suspected on the basis of clinical and laboratory findings and definitive diagnosis required liver biopsy and the diagnosis was confirmed by the finding of very low glucose-6-phosphatase activity [4,17].
Conclusion
GSD-1 should be considered in all children with unexplained hepatomegaly, with or without hypoglycemia. Orientation of young mothers about symptoms of GSD-1 to seek early medical advice is very important. Elevated serum biotinidase can be used as a diagnostic marker for hepatic glycogen storage disorders.
References
- Balistreri WF, Gray RG (2011) Metabolic diseases of the liver. In: Nelson Textbook of pediatrics 19th ed. Kliegman RM, Behrman RE, et al. (Eds.), Elsevier Saunders, USA, pp. 1388-1389.
- Priya SK, Yuan TC (2011) Defects in metabolism of carbohydrates. In: Nelson Textbook of pediatrics 19th (edn), Kliegman RM, et al. (Eds.), Elsevier Saunders, USA, pp. 492-501.
- Hendriksz J, Christian Gissen Paul (2010) Glycogen storage disease, Symposium: inborn errors of metabolism. Pediatrics and child health 25(2): 84-89.
- Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, et al. (2002) Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr 161(1): 20-34.
- Hicks J, Wartchow E, Mierau G (2011) Glycogen storage diseases: A brief review and update on clinical features, genetic abnormalities, pathologic features and treatment. Ultrastruct Pathol 35(5): 183-196.
- Wolf B, Freehauf CL, Thomas JA, Gordon PL, Greene CL, et al. (2003) Markedly elevated serum biotinidase activity may indicate glycogen storage disease type Ia. Journal of Inherited Metabolic Disease 26(8): 805-809.
- Paesold Burda P, Baumgartner MR, Santer R, Bosshard NU, Steinmann B (2007) Elevated serum biotinidase activity in hepatic glycogen storage disorders- A convenient biomarker. Journal of Inherited Metabolic Disease 30(6): 896-902.
- Koshy A, Ramaswamy K, Correa M, Rekha S (2006) Glycogen storage disease: report of 17 cases from southern India. Indian J Gastroenterol 25(4): 182-184.
- Koide H, Oda T (1959) Pathological occurrence of glucose-6- phosphatase in liver disease. Clin Chim Acta 4: 554-561.
- King J (1965) In: Practical clinical enzymology . D Van Nostrand Company Ltd, 1-301.
- Gogus S, Kocak N, Ciliv G, Karabulut E, Akcoren Z, et al. (2002) Histologic features of the liver in type Ia glycogen storage disease: comparative study between different age groups and consecutive biopsies. Pediatr Dev Pathol 5(3): 299-304.
- Shawky MR, Elsayed SN, Ibrahim SD, Seifeldin SN (2012) Profile of genetic disorders prevalent in northeast region of Cairo, Egypt. The Egyptian Journal of Medical Human Genetics 13(1): 45-62.
- Moraru E, Cuvinciuc O, Antonesei L, Mihãilã D, Bozomitu L, et al. (2007) Glycogen storage disease type-I between chronic ambulatory follow up and pediatric emergency. J Gastrointestin Liver Dis 16(1): 47-51.
- Eminoğlu TF, Tümer L, Okur I, Ezgü SF, Hasanoğlu A (2013) Clinical course and outcome of glycogen storage disease type 1a and type 1b. Turk Arch Ped 48: 117-122.
- Saltik N, Özen H, Ciliv G, Koçak N, Yüce A, et al. (2000) Glycogen storage disease type I a: frequency and clinical course in turkish children. Indian J Pediatr 67(7): 497-501.
- Kamolsilp M (2005) Glycogen storage diseases in thai patients: phramongkutklao hospital experience. J Med Assoc Thai 88(3): 295- 301.
- Hou J, Wang T (2003) A 20 year follow up of a male patient with type-Ia glycogen storage disease. Chang Gung Med J 26(4): 283-287.
- Carvalho PM, Silva NJ, Dias PG, Porto JE, Santos LC, et al. (2013) Glycogen storage disease type Ia- a secondary cause for hyperlipidemia: report of five cases. J Diabetes Metab Disord 12(25): 1-10.
- Balistrei WF, Carey RG (2011) Metabolic diseases of the liver. In: Nelson Textbook of pediatrics 19th (edn), Kliegman RM, et al. (Eds.), Elsevier Saunders, USA, pp. 1388-1404.
- Wolf B, Freehauf LC, Thomas AJ, Gordon LP, Greene LC, et al. (2003) Markedly elevated serum biotinidase activity may indicate glycogen storage disease type Ia. J Inher Metab Dis 26(8): 805-809.
- Angaroni CJ, Giner Ayala AN, Hill LP, Guelbert NB, Paschini Capra AE, et al. (2010) Evaluation of the biotinidase activity in hepatic glycogen storage disease patients. Undescribed genetic finding associated with atypical enzymatic behavior: an outlook. J Inherit Metab Dis 33(2): 289-294.
- Paesold Burda P, Baumgartner MR, Santer R, Bosshard NU, Steinmann B (2007) Elevated serum biotinidase activity in hepatic glycogen storage disorders-a convenient biomarker. J Inher Metab Dis 30(6): 896-902.















