




The Therapeutic Journey of Heterocyclic Compound 2, 4- Thiazolidinediones as A Versatile Pharmacophore: An Insight into Its Synthesis Routes, Structure Activity Relationships, & Biological Significance
Shubham Kenekar1* and Shital Chavan2
1Student, Department of Pharmaceutical Chemistry, Government College of Pharmacy, Aurangabad, Maharashtra , India
2Assistant Professor, Government College of Pharmacy, Aurangabad, Maharashtra, India
Submission: July 03, 2023;Published: July 11, 2023
*Corresponding author: Shubham Kenekar, Student, Department of Pharmaceutical Chemistry, Government College of Pharmacy, Aurangabad, Maharashtra , India
How to cite this article: Shubham Kenekar* and Shital Chavan. The Therapeutic Journey of Heterocyclic Compound 2, 4- Thiazolidinediones as A Versatile Pharmacophore: An Insight into Its Synthesis Routes, Structure Activity Relationships, & Biological Significance. Organic & Medicinal Chem IJ. 2023; 12(3): 555837. DOI: 10.19080/OMCIJ.2023.12.555837
Abstract
Thiazolidine 2,4dione is a pervasive heterocyclic pharmacophore with numerous pharmacological Significance and an immense chance for underlying change. The different scope of natural activities invested with an original method of synthesis, minimal expense, and simple blend has drawn in the consideration of restorative scientists. The 2,4-thiazolidinedione core has been combined with a variety of structural fragments to create a wide range of lead molecules that can be used to treat a variety of clinical conditions. At 2,4-TZD nucleus, the most frequently modified structural sites are N-3 and the active methylene at C-5. The article discusses the recent development of 2,4 thiazolidinedione’s derivatives as antimicrobial, anti-cancer, anti-diabetics, and other applications. The metabolic pathway governed by the peroxisome proliferator-activated receptor-gamma (PPAR-) is the target receptor with which 2,4 thiazolidinedione’s interacts most frequently. The 2,4-TZD core can also be synthesized using a variety of synthetic methods described in the article. Additionally, the mechanisms of action structure-activity relationships and the significance of various substitutions at N-3 and C-5 are also discussed. We hope that the scientific community working on the structural exploitation of the 2,4-TZD core for the creation of novel drug molecules for life-threatening conditions will find this article to be a useful resource.
Graphical Abstract: Figure 1
Background: Medicinal and pharmacological research serves as a foundation for creating novel treatment options for human disease. Chemistry is the science of the composition, structure, property, and reaction of matter, particularly of the atomic and molecular systems of all living organisms composed of numerous organic substances. The research obviates established therapy in favor of never metabolites, which are safer and more effective: Chemistry determines our identities, ancestry, and future. Utilizing several chemistry laws, pharmaceutical chemistry investigates drug preparation, chemical nature, composition, and structure’s impact on organisms, as well as the physical and chemical properties of drugs, their quality control procedures, and their storage conditions. Among the related sciences, pharmaceutical chemistry has held the most significant position. It is necessary to consider the physiochemical properties utilized in creating new pharmacologically active compounds and their mechanism of action to provide an understanding of the fundamentals of medicinal chemistry. However, several factors, including emerging infectious diseases and an increasing number of multi-drug-resistant microbial pathogens, particularly Gram-positive bacteria, make the treatment of infectious diseases an important and challenging issue.
The chemical and life sciences rely heavily on heterocyclic compounds. Our biological system relies heavily on heterocyclic compounds. A wide range of drug candidates, including antibiotic, anti-tumor, anti-inflammatory, antiviral, antimicrobial, antifungal, and anti-diabetic, contain heterocyclic moieties [1]. A wide range of pharmacological processes are carried out by heterocyclic moieties containing sulfur and nitrogen. Researchers who have synthesized and screened a variety of thiazolidinediones derivatives for their various biological activities found this to be of interest. We have tried to collect the biological properties of thiazolidinediones and their synthetic derivatives in this review [2].
Keywords: Thiazolidine 2,4 Dione; PPARγ; Molecular Modelling; Anti-Cancer; Anti-Inflammatory; Anti-Fungal; Antioxidant; Anti-Diabetic
Abbreviations: TZD: Thiazolidine 2,4 Dione; MOA: Mechanism Of Action; SAR: Structural Activity Relationship; USFDA: United States Food & Drugs Administration; MCF-7: Michigan Cancer Foundation-7; MDA-MDB: M. D. Anderson-Metastatic Breast Cancer Cell Line; MDA-MB 231: M. D. Anderson Metastatic Human Breast Cancer; MEK 1: Mitogen Activated Protein Kinase; PI3Ka: Phosphoinositide 3- Kinase Alpha; P-ERK: P- Extracellular Signal-Regulated Kinase; P- AKT : P- Ak Strain Transforming; VEGFR 2: Vascular Endothelium Growth Factor Regulator -2; HBA-HBD: Hydrogen Bond Acceptor- Hydrogen Bond Donor; NLRP-3: Nucleotide Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3; Irkβ: Insulin Receptor Kinase Beta; E.W.G: Electron Withdrawing Group; E.R.G: Electron Releasing Group; C. Albicans: Candida Albicans; S. Aureus: Staphylococcus Aureus; P. Aeruginosa: Pseudomonas Aeruginosa; E. Coli: Escherichia Coli; ROS: Reactive Oxygen Species ; DPPH: 2,2 Diphenyl Picrylhydrazyl; PPARγ: Peroxisome Proliferator Activated Receptor-Gamma; PTP1B: Protein Tyrosine Phosphatase 1B; BHT -Butylated Hydroxytoluene; ABTS; 2,2’-Azino-Bis (3-Ethylbenzothiazoline-6-Sulfonic Acids); STZ Induced Diabetic Rats: Streptococci Induced Diabetic Rat
Main Text
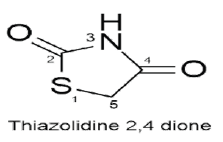
The heterocyclic ring systems known as thiazolidinediones were first developed toward the end of the 1990s to treat type 2 diabetes. TZD produce their biological action by stimulating the PPAR (Peroxisome proliferator-activated receptor), making the receptor active; PPAR is group of nuclear receptors that control the genes that regulate glucose homeostasis and lipid metabolism and are most specific for PPARγ (PPARα, PPARβ, and PPARγ are subtypes of PPAR). A thiazolidine ring with five members and two carbonyl groups makes up thiazolidinedione. The structure and properties of thiazolidinedione undergo the greatest change when variable substitutions are made at positions 3 and 5, respectively [3]. Rhodanine derivatives are produced when sulfur removes oxygen from position 2, which is evaluated for a variety of biological properties, including anti-cancer, anti-bacterial, antiaging, and anti-inflammatory properties [4]. Oxygen’s removes sulphur at position 1 produces oxazolidine Dione derivatives, which are commonly utilized as antibiotics. Linezolid was the first antibiotic in this class to receive FDA approval in 1999 [5]. The diverse biological functions of thiazolidine 2,4 Dione make it a potent heterocyclic ring. Researchers are currently continuously investigating this scaffold for the design and synthesis of novel compounds. The diverse and adaptable nature of the thiazolidine 2,4 diones moiety is the primary motivation for exploratory research.
Different possible targets for thiazolidine 2,4 diones scaffold:
Aldose Reductase (ALR2)
Phosphoinositide3-kinase γ
Pim Kinase 1
Peroxisome Proliferator-Activated Receptor (PPARγ)
Protein Tyrosine Phosphatase 1B (PTP1B)
Tyrosinase inhibitors
Mutant Thyroid Hormone Receptor-β (TR-β)
Phosphoinositide-3-kinase α (PI3Kα) and Mitogen-
Activated Protein Kinase (MEK)
Mutant Thyroid Hormone Receptor-β (TR-β)
UDP-N-acetylmuramoylalanine-D-glutamate Ligase (MurD Ligase)
Cyclooxygenase-2 (COX-2)
Histone Deacetylase 1
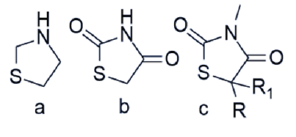
Structure & Biological Functions of PPAR:
PPARs are a group of receptors belonging to the family of nuclear retinoic acid receptors (RARs), thyroid hormone receptors, Steroid receptors which are involved in different biological processes. PPARs are a subfamily of transducer proteins that act as transcriptional factors. These receptors were first identified in rodents in the 1990s. Three major types of PPAR, encoded by separate genes, have been identified; they are PPAR-α (NR1C1), PPAR-β/δ (NR1C2), and PPAR-γ (NR1C3). All of the PPAR types are structural/ functional analogs of each other. The structure of PPAR has main 4 domains, knowns as A/B/C/D and E/F shown in figure [1] The domain A/B presented at the -NH2 terminal contains ligand-independent activation function 1 (AF-1) which leads the two phosphorylation of the PPAR. The domain C, also known as DNA-BINDING DOMAIN (DBD) comprises 2 zinc atoms that facilitate the binding of peroxisome proliferator response element (PPRE) to PPAR in the target gene promotor region. D domain works as a docking domain for the binding of coactivators to the DNA receptors. The remaining E/F domain is present near the AF-2, called as ligand binding domain (LBD) used to specify the ligand and activate the binding of PPAR to PPRE, thus promoting targeted gene expression & also promotes the recruitment of cofactors required for the gene transcription [6]. (Figure 2,3(a))
General Structure-Activity Relationship of 2,4 Tzd
The carbonyl group at 2 & 4 positions enhanced the activity of ring moiety.
The carbonyl group at 4 positions is highly unreactive, so replacement or substitution at the 4-carbonyl group will demolish the activity of the ring.
Substitution at 2, 3, and 5 -positions are possible.
Substitution at 5th position is most preferable.
Substitution of carbonyl oxygen with sulphur at 2nd position will result in new moiety rhodanine.
Replacement of sulfur at 1st position with oxygen forms oxazolidine diones which commonly utilized as anti-biotics.

Chemistry:
2,4-diones of 1,3-thiazolidine are thiazolidine derivatives has two carbonyl moieties at the second and fourth positions, leaving the thiazole ring’s -NH and methylene (-CH2) unaffected by structural modifications, allowing for the creation of numerous analogues. (Figure 3(b)) The biological activity of TZD derivatives can be altered by varying the substituents at positions three and five. (Figure 3(c)) Thiazolidinedione has a pKa of 6.82, according to reports [7].


Synthesis:
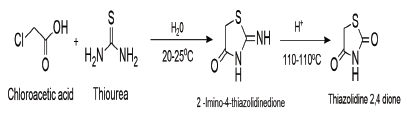
Thiocarbamates, thioureas, thiazolidine, alkali thiocyanates, and other starting materials are utilized in the synthesis of the thiazolidine 2,4 diones. The refluxing of chloroacetic acid and thiourea in the presence of water and HCL for eight to ten hours at 100- 110 °C is the most straightforward and conventional method for the synthesis of thiazolidine 2,4 diones. via the intermediate formation of 2-imino-4-thiazolidinedione as shown in scheme 1 [8].
Scheme 1
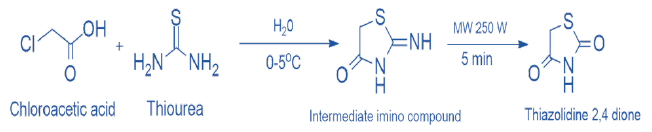
The above reaction can be further accelerated using a microwave (MW) assisted method, in this method, -chloroacetic acid is first reacted with thiourea in cold conditions (0-5°C) to produce 2-imino-4-thiazolidinedione, which is then irradiated with a microwave at 250W for 5 minutes to produce white crystals of thiazolidine 2,4 diones as shown in scheme 2 [9].
Scheme 2
Utilizing thiocarbamates as a starting material is yet another method for the production of thiazolidine 2,4 diones, as depicted in scheme 3 (9). Alkyl thiocarbamate can be made by reacting carbonyl sulfide with a primary amine in the presence of potassium hydroxide. The reaction of these alkyl thiocarbamates with halo alkanoic acids results in the formation of thiocarbamates, which cyclize to produce a TZD scaffold.
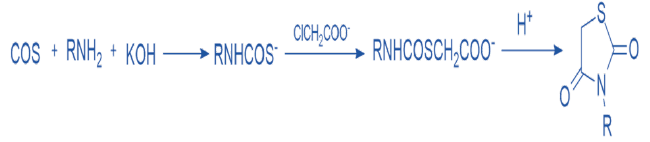
Scheme 3
One more way of synthesizing thiazolidine 2,4 diones is to react thiosemicarbazone in acetone with chloroacetic acid ester. This produces 2-hydrazino-4-thiazolidinedione in the presence of sodium ethoxide, which then produces a TZD nucleus in the presence of diluted hydrochloric acid [9].
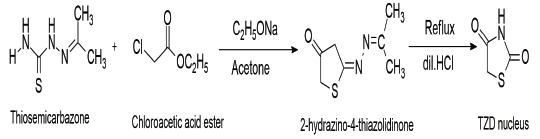
Scheme 4
Acidification (using dil. HCl) is an additional method for obtaining TZD scaffold of the product of the chemical reaction between potassium thiocyanate and ethyl chloroacetate. scheme 4 [9]. (Figure 4)
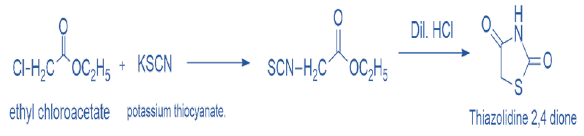

Tautomerism:
Figure 5 depicts the various tautomer obtained because thiazolidine-2,4-dione can undergo tautomerization due to its two carbonyl groups and hydrogen [10]. goes through either the amide-imidol or keto-enol type of tautomerism, or both types. There are several pharmaceutical products with the TZD ring that can undergo tautomerism, which typically involves protons migrating from one molecule site to another. It has been discovered that tautomer (a) is the most stable of the various tautomeric forms mentioned [10].

Structures of Different Tautomer’s:
(Figure 5)
Characterization:
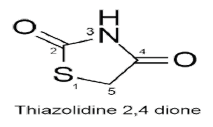
The unsubstituted TZD nucleus exists as a white crystalline solid with a melting point range of 120-122°C. The infrared (IR) spectrum of the same depicts an NH stretch at 3145cm-1, CH2 stretch at 2923cm-1, carbonyl stretch at 1738 cm-1 and 1659 cm-1 (for C2 and C4 carbonyls respectively), C-N stretch at 1318 cm-1 and 1165 cm-1, and C-S stretch at 808 cm-1 and 727 cm- 1[11]. The 1H-NMR depicts a singlet for CH2 protons at δ 3.98 (ppm) and a broad singlet for NH protons at δ 12.51 (ppm), which is due to the DE shielding effect of two carbonyl groups present on both sides [11] The mass spectrum of TZD depicts a base peak at m/z 116 (100%) [10].
Substitution on TZD Nucleus:
At positions 2 and 4, thiazolidine 2,4 diones has two carbonyl groups. However, since the carbonyl group at position 4 is extremely unreactive, researchers are interested in substituting at positions 2, 3, and 5. Substitution positions, or the free -NH and -CH2 moieties of the TZD core’s carbonyl group at two positions, have been investigated for the creation of numerous TZD derivatives.
Synthesis of 2-Substituted Thiazolidine 2,4 Diones:
It involves substituting acetophenone with thiourea and iodine at 60 °C to afford the intermediate, followed by the refluxing of chloroacetic chloride in dioxane respective 2-chloroacetamido substituted thiazoles & cyclized in the presence of ammonium thiocyanate to give 2-substituted thiazolidinedione derivatives [12].
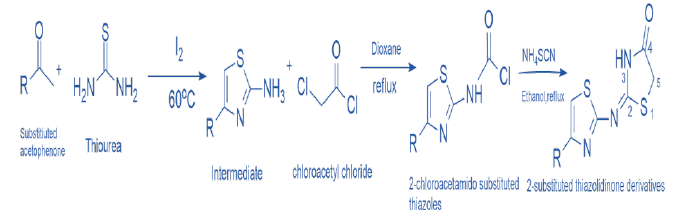
Scheme 5
Substitutions At -NH Moiety of TZD Nucleus:
The free -NH moiety of TZD is alkylated by using alkyl or aryl halides in the presence of alkali including sodium hydride [13] tetrabutylammonium iodide [14] or potassium carbonate [15] using DMF (N, N-Dimethylformamide) or acetone as a solvent.
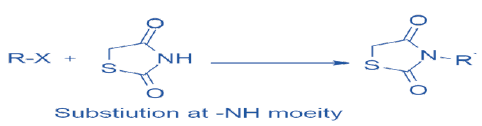
Scheme 6
Substitutions At Free -CH2 Moiety of TZD Nucleus:
The methylene moiety is substituted with aldehydes or ketones leading to the formation of arylidene derivatives, via ‘Knoevenagel condensation’. A Knoevenagel condensation is a nucleophilic addition type of reaction, which involves the addition of active hydrogen compound to a carbonyl group followed by a dehydration reaction, resulting in the loss of water molecule (hence condensation). The product formed is often termed an α, β-unsaturated ketone. Generally, occurs in the presence of a catalyst, usually a weakly basic amine. The condensation of TZD & Aldehyde is carried out under various reaction conditions including, a few drops of piperidine using ethanol or methanol as solvents for 7-42 hours [16] scheme 8 or anhydrous sodium acetate in glacial acetic acid while condensation of TZD with ketones has been carried out in the presence of ammonium acetate or piperidinium acetate in toluene or ethyl acetate [17] Also, different approaches are employed in the synthesis of this substituent like Green Synthesis which is mainly focused on eco-friendly, less hazardous chemical synthesis, developing a reaction condition for Knoevenagel condensation using Amino Acid L-tyrosine in water or β-alanine in acetic acid.
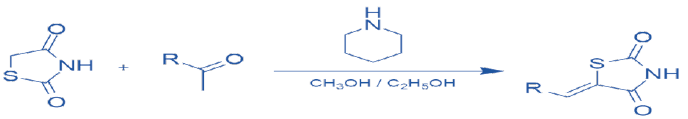
Scheme 7
(Figure 6)

Molecular Designing and Molecular Docking
Molecular designing provides a new approach to drug discovery. The traditional process of drug discovery & drug development is very tedious and economically costly. Combinatorial chemistry is a method for synthesizing many structurally distinct molecules at once and submitting them to high throughput screening (HTS) assays. One of the most recent approaches that have been developed by researchers working in the pharmaceutical sector is called combinatorial chemistry. Its goal is to cut down on the amount of time and money required to produce new drugs that are both effective and competitive. This method is having a significant impact on all areas of chemistry, particularly drug discovery, by accelerating the production of biologically active compounds. Compound libraries can now be created to search for novel bioactivities through combinatorial chemistry’s rapid advancements. Pharma companies are beginning to benefit from the rapid discovery of novel drug candidates, significant savings in preclinical development costs, and ultimately a shift in their fundamental approach to drug discovery thanks to this potent new technology. The inventive process of finding new medications based on knowledge of a biological target is known as drug design. It entails making molecules that are compatible with the shape and charge of the biomolecular target with which they will interact and bind. The Traditional life cycle of drug design described in Figure 7 & modern drug design process depicted in Figure 8.

Modern Drug Design:
(Figure 9)
Drug design with the help of computers may be used at any of the drug discovery stages like Hit identification using virtual screening (structure or by ligand-based design), Hit to lead optimization of affinity and selectivity (structure-based design, QSAR), Lead optimization: optimization of other pharmaceutical properties’ while max affinity and min. toxicity.
Objectives of Computational Chemistry:
Speed up screening processes.
Efficient screening (focused, target-directed)
De novo design (target directed)
Integration of testing into the design process
Fail drugs remove fast.
Ligand-Based Drug Design is based on knowing which other molecules bind to the desired biological target. Used to derive a pharmacophore model that defines the minimum necessary structural characteristics a molecule must possess to bind to the target. LBDD offers a general approach for elucidating relationships between the structural and physiochemical properties of compounds/ligands and their biological activities. Alternatively, a QSAR in which a correlation between calculated properties of molecules and their experimentally determined biological activity, may be derived. These QSAR relationships in turn may be used to predict the activity of new analogs. Shown in (Figure 9).


Structure-Based Drug Design relies on knowledge of the three-dimensional structure of the biological target obtained through X-Ray crystallography or NMR Spectroscopy. Using the structure of the biological target, candidate drugs that are predicted to bind with high affinity and selectivity to the target may be designed using, interactive graphics and Various computational procedures.
Methods
Virtual Screening: The first method is the identification of new ligands for a given receptor by searching a large database of small molecules to find those fitting the binding pocket of the receptor.
De Novo Design of New Ligands: In this method, ligand molecules are built up within the constraints of the binding pocket by assembling small pieces in a stepwise manner. The key advantage of such a method is that novel structures can be suggested.
Optimization of known ligands: By evaluating proposed analogs within the binding cavity.
Steps:
Binding Site Identification: the first step in structured-based design. Relies on the identification of concave surfaces on the protein that can accommodate drug-sized molecules that also app. Hot spots {hydrophobic surfaces, hydrogen binding sites} that drive the ligand binding.
Docking and Scoring: best matching between two molecules. Includes lock and key model. Right key for the lock. To place a ligand (small molecules) into the binding site of a receptor in the manners appropriate for optimal interactions with a receptor.
A variety of computational tools are used to design a molecule target, also known as a molecular ligand, and to estimate how well it will dock with a target receptor by interpreting various parameters. The databases that are used to refer to the drug target or, for example, the reference ligand drug molecule protein database, including PDB0, zinc databases, zinc15Database, ChEMBL, and J Chem for Excel. Chem Draw, Marvin sketch, ACD/ Che sketch, Marvin molecular editor and viewer, pymol, and chem writer are among the drawing tools that can be used to draw the structure of promising drug candidates. CHARMM, GROMICS, AMBER, CHARMM- GUI, and Swiss side chain are among the drug design-related applications of molecular modelling tools. The software used to predict the binding site are MED-SuMo, CAVER, FIND SITE, and Pocket Annotate database. To determine whether the tested compound successfully docks in the target receptor’s docking site of search compound and targe, Schrodinger, iGemdo, Auto dock, DOCK, GOLD, and Swiss Dock are used. Target prediction and QSAR studies MolScore -antivirals, MolScore antibiotics, SEA, Chemprot, and Cqsar, clogs, MOLEdb, them DB, OCHEM, Pattern Match Counter, and Avogadro used respectively. Vol Surf, Gastropub, Med Chem Studio, AGPS, and Metabases are utilized for toxicity studies.
Biological Significance of Thiazolidine 2,4 Diones:
Anti-Cancer Agents:
Since cancer was first identified as a disease, its enigmatic origins and even more enigmatic mechanisms of spread have always piqued human curiosity. Over the years, researchers worldwide have been diligently attempting to reveal the mysteries of cancer. For the research and development of anticancer drugs, this is a very exciting time, one that has a lot of opportunities and challenges. Researchers are always looking for new anticancer drugs, and over the past 50 years, a lot of effort has been put into making anticancer drugs that work better. There are some significant new directions that the 21st century holds. The concept of systemic chemotherapy, which is specific for cancer cells and does not cause major side effects, has had a significant impact on cancer treatment, despite advancements in radiotherapy and surgery remaining an important objective for the future. From an understanding of how cancer growth is controlled to the technology of drug synthesis and testing to the multifactorial requirements for clinical trials, the issues underlying the achievement of this goal are complex. Even though numerous chemotherapeutic treatments for cancer have been proposed, tried, and in some cases implemented over the past few decades, these diseases continue to be fatal and persistent. To combat this disease, new treatments with novel mechanisms are therefore urgently required. So, in the early 2000s, much research had been done on thiazolidine ring, as a potential anticancer scaffold. They found that among the other heterocyclic scaffolds; Thiazolidinediones play an effective role in the prevention of cancer development & metastasis. Many mechanisms are involved in cancer progression out of which Raf/MEK/ERK (extracellular signal-regulated kinase), PI3K (phosphatidylinositol 3-kinase), Went signal transduction pathways, and PPARs (peroxisome proliferator-activated receptors) are the most commonly upregulated signaling pathways in human cancers [6] (Figure 10).
Salamone et.al reported new derivatives of troglitazone, which was mainly used as an anti-diabetic agent & found that they exhibit effective anti-proliferative activity on various cancer cell lines. Newly synthesized derivatives of troglitazone were assayed for their anti-proliferative activity against hormone-dependent (MCF-7) & Hormone independent (MDA-MB-231) breast cancer cell lines. They concluded that TGZ derivatives are more reactive towards the hormone-independent cell line (MDA-MB-231) than the hormone-dependent cell line (MCF-7). SAR studies show that the double bond influences the activity, unsaturated derivatives are more active than saturated derivatives against both cell
lines. Also, major problems in thiazolidinediones are their hepatotoxicity. Compound 1 suggests that lipophilic substituents on the chromone ring exhibit much less toxicity than hepatocytes. Compound 2 is devoid of a hydroxyl group and unable to be transformed into toxic metabolites. Compound 2 showed significant results against hormone-dependent (MCF-7) and hormone-independent (MDAMDB- 231) breast cancer cell lines with IC50 of 12.5 ± 0.9 & 10.8 ± 0.37 with 84% hepatocyte viability using troglitazone as reference drug. Compound 1 also showed promising results against hormone-dependent (MCF-7) and hormone-independent (MDA-MDB-231) breast cancer cell lines with IC50 of 13.0 ± 1.5 & 3.2 ± 0.3 respectively with 79% hepatocyte viability using troglitazone as the reference drug [18].


K. Liu et.al synthesizes a series of 3,5 di substituted TZD to evaluate their anti-cancer properties. They found potent inhibitory effects on the growth of the U937 cell line which is the pro-monocytic human myeloid leukaemia cell line. After a series of derivatization and their evaluation SAR studies show that for anti-cancer activity:
Exocyclic double bond: Essential for anti-cancer activity.
Aromatic/ Phenyl ring: the presence of an aromatic ring is also essential to execute the anti-cancer activity, replacement of a phenyl ring will decrease potency/activity. Whereas substitution of the aromatic ring with another ring like a cyclopropane ring will greatly influence the activity & substitution of the aromatic rings with EWG (NO2) will decrease its growth inhibition. Substitution with EDG (OCH3) will result in comparable potency. Substitution of aromatic ring with cyclohexane ring will increase the potency by 3 times fold due to the important role of steric effect at its specific domain.
Ethylamine domain: Important for good anti-cancer activity. Replacement with carboxy/ that. The amino group significantly decreased in activity, total loss of activity. Replacement of -NH2 with -OH (CH3OH) maintained biological activity in U937 human leukaemia cells this suggests that ionic interaction with certain acidic amino acid residues in target protein structure is important for biological activity while steric effects are not favored at this specific site.
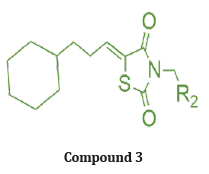
Out of many synthesized compounds, compound (3) docked into the ATP binding pockets of MEK1 and PI3K. The docking results suggest that the lead compound fits nicely into the ATP binding pocket of both MEK1 and PI3K. As shown in (Figure 11) interacts with the MEK1 through H-bond interactions of the ethylamine with the Asp190, Asn195, and Asp208 residues. The two carbonyl groups of TZD also interact with Lys97 and Ser194 through H-bonds. The cyclohexane ring of compound 3 fits into a hydrophobic pocket. Similarly, ethylamine interacts with Glu768 and Glu798 of PI3K through H-bond interactions. The tested compound shows enhanced growth inhibition in less than 3 μM indicating signaling blockage and growth inhibition in U937 cells. & also tested in androgen-insensitive prostate PC-3, DU145, and M12 cells to evaluate the anti-proliferative and signaling cascade inhibitory effects. More importantly, consistently inhibited the PI3K/Akt and Raf/MEK/ERK signaling pathways as seen in the suppressed level of p- ERK and p-Akt in DU145 cells [19].
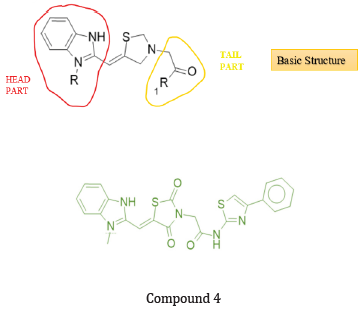
P. sharma et.al prepared a series of new benzimidazolelinked TZD hybrids & evaluated them for their cytotoxic potential against selected human cancer cell lines of the prostate (PC 3 and DU-145), breast (MDA-MB-231), lung (A549) and a normal breast epithelial cells (MCF10A) using 3-(4,5- dimethylthiazol-2- yl)-2,5-diphenyltetrazolium bromide (MTT) assay and 5 FU as a reference compound. Among the tested compounds, compound (4) exhibited promising cytotoxicity with an IC50 value of 11.46 ± 1.46 μM on the A549 lung cancer cell line but did not show significant toxicity on normal MCF10A cells having IC50 value > 100 μM. also exhibiting good anti-cancer activity on PC 3 (Prostate cancer cell line), DU-145 With IC50 values 39.87 ± 3.16μM & 31.41 ± 1.52 μM respectively. Compound (4) screened for activity on the MDA-MB-231 Breast cancer cell line had an IC50 value of 29.18 ± 1.24 μM. also treated with A549 Cells showed typical apoptotic morphology like cell shrinkage, chromatin condensation, and horseshoe-shaped nuclei formation. From data obtained from screened compounds, we can conclude that the N-substitution on the tail position of thiazolidinedione with oxoethyl-linked morpholine, piperidine, pyrrolidine, and 2,6-dimethylmorpholine does not form any biologically active compound. However, most of the compounds containing 6,7-dimethoxytetrahydroisoquinoline at the tail side of thiazolidinedione were found to be active (below 50 μM). On the other hand, the substitution of the head position of TZD with 1-methyl-benzimidazole produced more active compounds compared to the ethyl and isopropyl group [20].
Khaled EL ADL et.al made a series of novel benzylidene TZD hybrids & evaluated them against some cell lines; HepG2 (hepatocellular carcinoma), HCT -116 (colorectal carcinoma), MCF 7 cells (cell line of breast cancer) taking sorafenib as a reference drug molecule. Vascular endothelial growth factor receptor -2 (VEGFR 2) is a receptor for VEGF & prime mediator of VEGF-induced pro-angiogenesis signaling. The binding of VEGF TO VEGFR-2 leads to the dimerization of receptors, the action of tyrosine kinase, trans autophosphorylation & initiation of extracellular signal-regulated kinase. Angiogenesis is important, a significant event in tumor development, so blocking angiogenesis is one of the most promising strategies to treat malignancies.
Based on Their Study SAR Of VEGFR2 Inhibitors Shared Main 4 Features:
• Flat heterocyclic ring that contains at least one nitrogen atom.
• A central aryl ring {hydrophobic spacer}
• A linker containing a functional group as a pharmacophore (NH3 / urea that possesses both an H bond acceptor & H bond donor to bind 2 crucial residues (Glu 883 / Asp 1044) of the target protein.
• Terminal hydrophobic moiety- essential for occupying allosteric hydrophobic pocket.
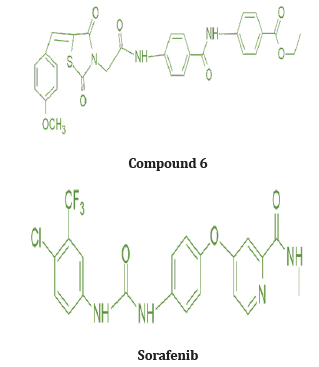
They replaced pyridine & 5 Fluro-2- oxoindolin-3-ylidene moieties from the reference drug molecule i.e., Sorafenib & Sunitinib with 4 chlorobenzyl Dene or 2,4 dichlorobenzylidene serves as a hydrophobic portion. Moreover, the thiazolidine 2,4 diones motif was designed to replace the central aryl ring and 5-member pyrrole ring of reference ligands (Sorafenib & Sutinib) respectively. It forms 5-(4-chloro benzylidene) thiazolidine 2,4 Dione or 5-(2,4 dichlorvos benzylidene) thiazolidine 2,4 Dione scaffolds linked to substituted hydrophobic phenyl tail through acetamide linkers containing HBA - HBD, interacts as HBA through its C=O & HBD through NH with essential Amino acid residues Asp 1044 & Glu 883. So based on that they synthesize series of 4 chloro, 2,4 dichloro derivatives compounds, by substituting on hydrophobic phenyl tail.
General SAR
• Spacers or linkers such as (Hydrogen bond donor & Hydrogen bond acceptors) lipophilicity & electronic nature are required for good anti-cancer activity.
• Linkers such as acetamide and carboxamide linkers were found to be higher in activity.
• Lipophilicity increases -Anti-cancer activity enhances. 2,4 dichlorobenzylidene derivative exhibit higher anti-cancer activities than 4 chloro substituent, Due to an increase in the lipophilic nature of substituents.
• Compounds with distal phenyl moieties show higher activities than distal aliphatic ones.
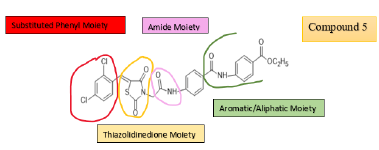
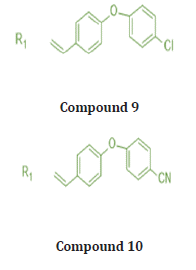
Out of many compounds, compound (5) (5‐([4‐Chloro/2,4‐ dichloro] benzylidene) thiazolidine‐2,4‐dione) which has 2,4 dichloro benzylidene & distal phenyl ring substituted with ethyl ester group shows the highest activities which is proved by docking studies, the proposed binding mode of compound (5) is virtually the same as that of standard sorafenib. Docking studies revealed the binding affinity value of compound (5) is -101.17 kcal/mol as compared to standard which was -95.36 kcal/mol. Figure 12, 13 Also, it shows prominent cytotoxic in vivo activity towards cell line HEPG2, HCT 116, MCF -17 with IC50 values of 9.16+0.9, 8.98+0.7, 5.49+0.5 micro m. Nearly similar to the reference ligand, in case of HepG2 & slightly lower in HCT 116 & higher activity in MCF -17 cancer cell line. The prominent test compound also tested for VEGFR 2 inhibitory activity, having an IC50 value of 0.17+0.02 μM, which is nearly more than half of the IC50 value of sorafenib (0.10+0.02 μM). So, compound (5) evolved as potential lead [21].
Considering the previous study, Khaled et.al synthesizes a new series with a substitution with more lipophilic nature and strong electronic nature. They replaced the chloro substitute with a methoxy group. The Methoxy group is a Strong electronegative atom with the -I effect, which pulls the electrons towards itself. In 4 methoxy benzylidene, Hydrogen bonding increases result in an increase in hydrophobic interactions ultimately affinity of the compound towards receptors is enhanced. All the modelling experiments were performed using MuleSoft software. The structure of VEGFR-2 was downloaded from Brookhaven protein data bank (PDB ID: 1YWN) Compared to standard sorafenib, urea linker formed 1 H bond with key amino acid Glu883 through its NH group and one H bond with Asp 1044 through its carbonyl group, which is responsible for the higher affinity of sorafenib towards VEGFR receptors. The Lipophilic substituent 4 methoxy benzylidene moiety on the aromatic hydrophobic ring occupied the hydrophobic groove formed by Arg1025, His1024, Ille1023, Cys1022, Leu1027, Ile890, Ile886 with more affinity. 4 methoxy benzylidene moiety compensated the effect of N methylpicolinamide moiety of sorafenib and increased hydrogen bonding and hydrophobic interactions and consequently affinities towards VEGFR -2 enzyme. compound (6) shows the highest binding affinity value of 103.50 kcal/mol more than that of std sorafenib shown in (Figure 14,15) Cytotoxic evaluation for Antiproliferative activity of newly synthesized methoxy benzylidene derivatives of thiazolidine-3,4 Dione was examined against 3 human tumor cell lines, namely HepG2, HCT 116, MCF-7, using MTT colorimetric assay. compound (6) with IC50 Values 6.19+0.5 μM, 5.47+0.3 μM, 7.26+0.3 μM for HepG2, HCT 116, MCF-7 cells respectively, which are higher than standard for HepG2, MCF-7 but lower activity towards HCT 116 cancer cell line. Also, compound (6) was found to be the most potent derivative that inhibits VEGFR 2 at an IC 50 value of 0.12 + 0.02 μM [22].
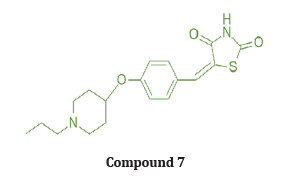
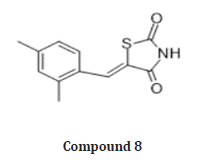
Vijay Laxmi synthesized novel 5-(4-alkylbenzyledene) thiazolidine 2,4 Dione derivatives by Knoevenagel condensation of 4 hydroxy benzaldehyde with TZD & evaluated for anti-cancer, antimicrobial activities using DNA cleavage studies. In vitro studies are Carried out on 60 human cancer cell lines, organized into nine sub-panels derived from 9 different human cancer cell types: Leukaemia, Lungs, Colon, CNS, Melanoma, Ovarian, Renal, Prostate, Breast cancer cell lines. among all the screened compounds, compound (7) shows significant growth inhibition against variety of cell lines at a single dose of 10-5 M concentration, exhibiting a broad spectrum of growth inhibition activity against all 9 tumor cell line types [23]. (Table 1)
Anti Inflammatory
Inflammation is a protective response involving immune cells, blood vessels, and molecular mediators that is part of the complex biological response of body tissues to harmful stimuli like pathogens, damaged cells, or irritants. Inflammation purges necrotic cells and tissues damaged by the initial insult and inflammatory process, initiates tissue repair, and removes the initial cause of cell injury: acute and chronic are the two main types of inflammation. The body’s first response to harmful stimuli is acute inflammation, which is caused by the increased movement of plasma and leukocytes-particularly granulocytes-from the blood into the injured tissues. A progression of biochemical occasions spreads and develops the provocative reaction, including the neighborhood vascular framework, the invulnerable framework, and different cells inside the harmed tissue. Chronic inflammation, also known as prolonged inflammation, is characterized by the simultaneous destruction and healing of the tissue caused by the inflammatory process and results in a progressive shift in the type of cells that are present at the site of the inflammation, such as mononuclear cells. Aggravation has additionally been delegated Type 1 and Type 2 in light of the sort of cytokines and assistant Lymphocytes (Th1 and Th2) involved [24]. (Figure 16)
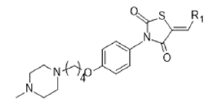
A Series of Novel benzo indole-2-one scaffolds containing thiazolidine 2, 4 Dione were synthesized by Mohd. Abdullah et.al evaluated them for their anti-inflammatory activity against new class of NLRP 3 Inflammasome inhibitors. The NLRP 3 Inflammasome is a critical component of innate immune system that mediates caspase-1- activation & the secretion of pro inflammatory cytokines IL-1B/ IL-18 in response to any microbial infection and cellular damage. Based on the previously proposed hypothesis of ER beta (estrogen receptor) beta agonist suppress the expression of NLRP 3 Inflammasome and other pro inflammatory markers TNF alpha, CD68, IL - 1 beta. They prepared about 20,000 libraries of compound using the target-based drug design, virtual screening against the crystal structure of ER beta and selected the top 10 compounds with best binding score and evaluated invitro for their anti-inflammatory activity against mouse macrophages (J774.1 cells) of NLRP 3 Inflammasome. benzylidene- thiazolidine-2,4 diones docked against ER beta protein and compound 8 shows a prominent glide score of -10.2 kcal/ mol and also docked with the ATP binding site of NLRP 3 protein with glide score of -9.1 Kcal/mol. The result of the anti- Inflammatory evaluation revealed that compound 8 shows superior inhibition of NLRP Inflammasome inhibition [25]. (Table 2)


Ahmed Elkamhawy synthesized some novel 3,5 substituted benzylidene piperazine substituted thiazolidinedione derivatives and evaluated them for IK beta modulator. Inhibition of IKK-β (inhibitor of nuclear factor kappa-B kinase subunit beta) has been broadly documented as a promising approach for the treatment of acute and chronic inflammatory diseases, cancer, and autoimmune diseases. Optimized their inhibitory activity by Insilco docking simulation study. They prepared a total of 18 new derivatives, out of which compound 9 found to be most potent inhibitory active agent with an IC50 Value of 0.26 ± 0.02.
Structure-Activity Relationship
(Table 3)
The SAR studies revealed that unfused aromatic ring is necessary to be attached with exomethylene carbon to obtain IKK β inhibitory action. Also suggests that the substitution at heterocyclic ring attached with unfused aromatic ring should be at para position, if it is at meta position, it will create a steric hindrance, is not favorable towards the binding site of IKK β protein [26]. (Figure 17,18) (Table 4)


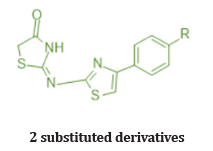
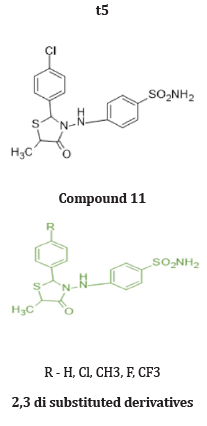
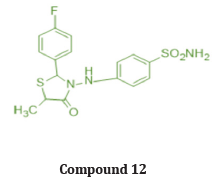
Abdullatif et.al prepared a 2 series of 2 substituted phenyl thiazole thiazolidine 4 one and 2,3,5 substituted derivatives. And evaluated for their invitro COX-2 selectively and Anti-inflammatory activity in vivo. Compound 11 and 12 exhibited COX-2 Inhibitory activity invitro comparable to celecoxib & showed in vivo antiinflammatory activity (edema inhibition -61.8% & 67% after 3 hr. respectively for compound 11 and 12), higher than that of reference standard drug celecoxib (60% after 3 hr.) at 10 mg/Kg dose employing on carrageenan induced pow edema method in mice. Based on their study, we can conclude that 3 substituted derivatives are more potent than 2 substituted derivatives for edema inhibition. Substitution at 4th position of 2-aryl ring increased the inhibition of selectively for COX-2 enzyme. Also, with EWG (Cl, CH3, -F) increases potency [27]. (Table 5)





Anti-Fungal
Fungi that are prevalent in the environment frequently result in fungal diseases. Pathogenic fungi can easily infect the body and cause disease if the immune system is weak, or the body’s flora is out of balance. An antifungal medication that works by selectively attacking the essential molecules of the structure of the fungus or key enzymes that are involved in the metabolic activity of the fungus. This stops the fungus from growing and has little or no effect on the mammalian host. However, it is challenging to develop antifungal medications because fungi, like humans, are eukaryotic organisms whose cells primarily consist of the cell membrane, cell wall, and cell nucleus. The molecular targets of the antifungal agents that are currently available can be used to classify anti-fungal agents. Enzymes and other molecules involved in the cell wall and plasma membrane synthesis, fungal DNA and protein synthesis, cellular function-related factors, and virulence factors are the primary molecular targets of antifungal agents [28]. (Figure 19)

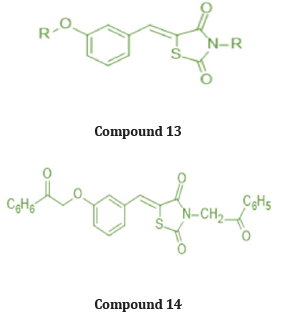
A series of new 3,5 disubstituted thiazolidine 2,4 diones was prepared by Gabriel Marc et.al by microwave-assisted synthesis. They evaluated all new compounds for their anti-fungal activity invitro testing performed on candida albicans ATCC10231 & used fluconazole as a reference drug. They performed the molecular docking with the help of auto dock 4.2, used fungal lanosterol 14α demethylase experimental structure isolated from C. Albicans as target molecule. The compound 14 was found to be most active with best binding free energy (-12.44) Kcal/mol as compared with reference drug with (-6.71) Kcal/mol, top docking conformation for compound 14 shown Figure 20 Molecular docking studies suggest that substitution on parent molecule (compound 13) with disubstituted carboxylic acids or large residues, significantly increased inhibitory potential (compound 14). Substitution at R position of parent molecule with aromatic rings are preferred than aliphatic/ alkyl substitutions. The carbonyl group (C=O) at substituted 3rd and 5th position are essential for activity forms H bonds with Ser 378 residue. Also, for better ADME -lipophilic substitution like phenyl, carbonyl, methyl (compound 14) or ester group are essential for better LogP values than the compound with polar groups (amide, carbonyl). Determined the Minimum inhibitory concentration and min. fungicidal concentration of best candidates through the screening assay using the diflumetorim method [29]. (Table 6-8)


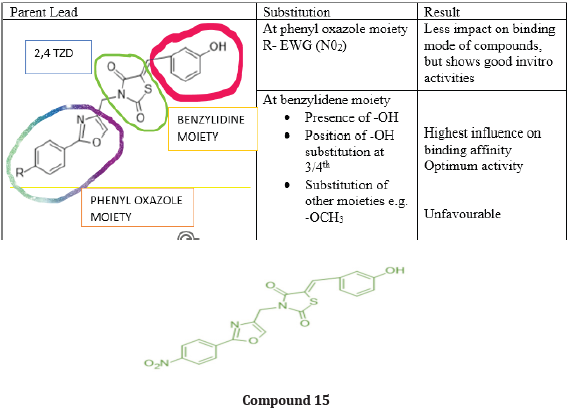
Gabriel Marc et.al synthesized some novel N-(oxazolyl methyl-1)- thiazolidine diones derivatives & evaluated them for antifungal and anti-microbial activities. Nearly all tested compounds show significant zone of inhibition in a range of (12-16 mm) which is mediocre in comparison with standard fluconazole (24 mm). They performed the molecular docking of active compounds, docked with all surface proteins of C. Albicans to find out the MOA of compounds & their binding affinity Compound 15 shows overall satisfactory docking results with all 9 Als targets & especially against Als 3, which is most important protein involved in biofilm development with binding energy of -10.81Kcal/mol shown in table (9) They performed invitro testing of biofilm activity of all new 16 compounds, out of which 14 were found to be active against biofilm formation at minimal biofilm eradication concentration (MBEC) smaller than standard berberine. The most active compound 15, maintained anti-biofilm activity even at concentration of 0.038 mg/mL, which is less than standard with 0.312 mg/ml. data shown in Table (8) [30] (Figure 21).




IC50 value represents the concentration of the compound required to produce 50% inhibition of COX-1 or COX-2 ED50 value used to represent anti-inflammatory activity which was the effective dose calculated after 3h.



Structure-Activity Relationships:
Antioxidant:
Antioxidants are substances that significantly delay or prevent the oxidation of an oxidizable substrate when present in relatively low concentrations. Antioxidants play a crucial role in intercepting and reacting with free radicals to prevent the cascade effect of ROS propagation by readily donating their proton to ROS (Reactive Oxygen Species) [31,32]. (Figure 22)

Harsh Kumar et. Al synthesized a 5- substituted (aryl/alkyl) methyl imino benzylidene 2,4 TZD derivatives & evaluated for their antioxidant activity. They prepared a total of 20 derivatives & all of them were assayed by applying DPPH (2,2 diphenyl-1- picrylhydrazyl) radical scavenging method using ascorbic acid as a standard drug. DPPH radical reacts with appropriate reducing agent to produce a new bond, result into change in color of the solution. As concentration of antioxidant increased, the more radicals taken up by the DPPH radical from antioxidant molecule that leads to loss in intensity of color of solution. This reactivity is taken to test the ability of compounds that can act as free radical scavengers. The IC50 value calculated for all the prepared compounds, out of which compound 16 found to be most active shows prominent activity as compared to standard. (Table 10, 11) [33].
Structure Activity Relationship:
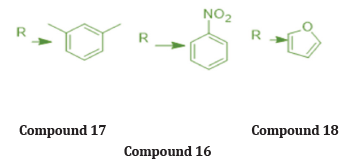
R = substituted phenyl moiety, substitution at O/P position with EDG (CH3) -increased anti-bacterial potential (compound 17)
R= Substitution with EWG at meta position (NO2) -Enhanced anti-bacterial & anti-fungal activity (compound 18)
R = replace phenyl ring with heterocyclic ring (furfuryl group) -result into potential antioxidant agent (compound 16)
Sucheta et.al synthesized a series of novels 5-substituted benzaldehyde thiazolidine 2,4 diones & evaluated them for antioxidant activity. Invitro antioxidant activity were assayed by applying a DPPH radical scavenging method & taking ascorbic acid as a standard molecule. Compound 19 and compound 20 were found to be the most potent among all the derivatives & possess a good antioxidant activity [34]. (Table 12)
Structure Activity Relationship:
Marc et.al synthesized a new series of (E)-5-(2-(5-(substituted benzylidene)-2,4-dioxothiazolidin-3-yl) acetyl)- 2-hydroxy benzamide & evaluated them for their invitro antioxidant property using ABTS & DPPH radical scavenging assays. When compared to Trolox (6-hydroxy-2,5,7,8-tetramethyl chroman 2-carboxylic acid), ascorbic acid, and butylated hydroxytoluene (BHT) as the standard compounds, the antioxidant activity tests revealed that all of the compounds exhibited moderate to potent radical scavenging activity. In addition, the activity of compounds 21 and compound 22 was even higher than that of the reference compounds [35]. (Table 13)
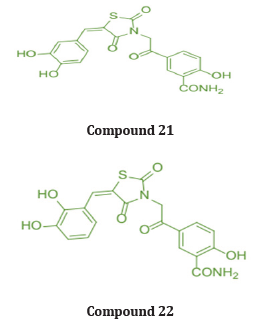
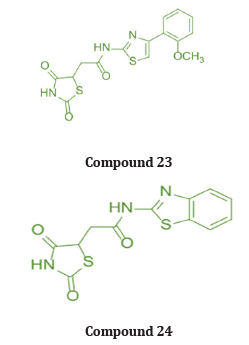
Koppireddi et.al used superoxide anion scavenging activity, DPPH radical scavenging activity, lipid peroxidation inhibition, and erythrocyte hemolysis inhibition assays, & synthesized a series of various TZD acetamide analogues fused with thiazole and benzothiazole scaffolds and assessed their antioxidant potential. When compared to standard ascorbic acid and luteolin, the activity of compound 23 and compound 24 were found to be moderate [36]. (Table 14)






Anti-Diabetic Activity
Diabetes is a chronic metabolic disorder that is linked to insulin resistance in the peripheral blood and impaired insulin secretion. It consists of a variety of dysfunctions, the first of which is hyperglycemia, which can lead to vascular diseases, nephropathy, vascular neuropathy, cardiovascular diseases, and other conditions if left untreated. Sorted into Types I and II; Eighty percent of the world’s affected population has Type II, which can be treated with lifestyle changes and the right medications. Type I diabetes is known as juvenile diabetes or insulin-dependent diabetes is a chronic condition in which the body’s insulin produces little or no insulin. Type II diabetes is the most common type, known as non-insulin-dependent diabetes usually in adults which occurs when the body becomes resistant to insulin or failed to produce insulin in sufficient amounts. According to WHO reports, about 422 million people worldwide have diabetes, and 1.5 million deaths are directly attributed to diabetes each year [37].
TZD commonly known as glitazones were first introduced in late 1990s to treat type II diabetes disorder, act through activation of PPARs a group of nuclear receptors that regulated genes controlling glucose homeostasis & lipid metabolism. They act either by trans-activation or by trans-repression mechanism, Shown in Figure 20, 21. The first drug candidate or prototype of TZD class was pioglitazone developed in late 1980s but due to severe liver toxicity never used as a medication. Late in 1998, a drug molecule named troglitazone was developed but it was also withdrawn from the market due to hepatotoxicity in the year 2000. In 1999 different TZD containing drug molecules were developed known as pioglitazone, englitazone, rosiglitazone, darglitazone. Out of which pioglitazone reported safe to use in hepatic complications & currently in use as anti-diabetic agent. The use of Rosiglitazone was restricted by the US FDA due to heart failure complications, but in the year 2013 the restrictions were removed by the FDA after series of trails. Pioglitazone and rosiglitazone’s are only 2 drugs which are currently available in market. (Figure 23) [38].

Mechanism of action as an Anti-diabetic agent


(Figure 23-25)
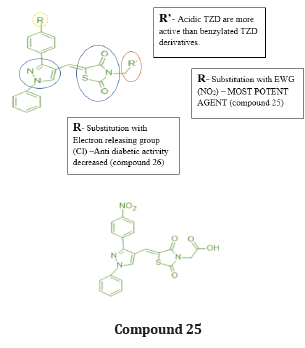
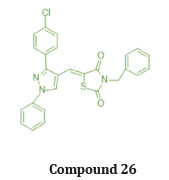
Bansal et.al synthesized a series of 14 novel TZD derivatives fused with pyrazole moiety. The synthesized compounds were docked against PPARγ & α amylase receptor (Figure 26, 27) & further evaluated for their invitro, in vivo anti-diabetic activity. Among all 14 compounds, compound 25 found to be most active with IC50 value in a range of 4.08 μg/mL when referred to standard ascorbic acid & shows significant blood glucose lowering action with active inhibition of alpha amylase. shown in (Table 14, 15) [39].


Structure activity relationship:
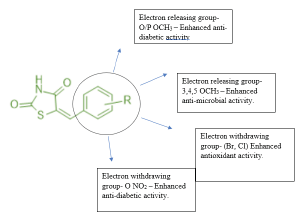
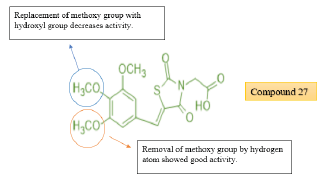
Datar et.al prepared a novel series of carboxylic ester derivatives of thiazolidine 2,4 diones substitution at N3 position and 5- substituted benzylidene. Among all the synthesized compounds, compound 27 was found to be the most potent derivative with a consistent decrease in the blood glucose level to 104 mg/dl for 120 min against pioglitazone as the standard drug molecule with 115 mg/dl [40]. (Table 15,16)

Structure activity relationship:
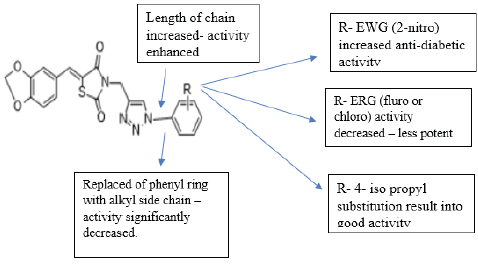
A new series of 1,2,3 triazoles tagged with TZD’s were synthesized by chinthala et.al. They evaluated them for antidiabetic activity profile, activity was assayed by invitro testing against α glucosidase enzyme. α glucosidase enzyme is an enzyme which delays carbohydrate digestion, causing reduction in rate of glucose & insulin level helps in treating hyperglycemic condition. Molecular docking studies were performed to analyze the conformation of synthesized compound, docking conformation of compounds shown in (figure 28 (a &b)) & also to evaluate inhibition of α glucosidase enzyme, results of docking studies shown in table 16. The best docking score compound further taken for invitro assessment (Table 17) [38].
Structure activity relationship:



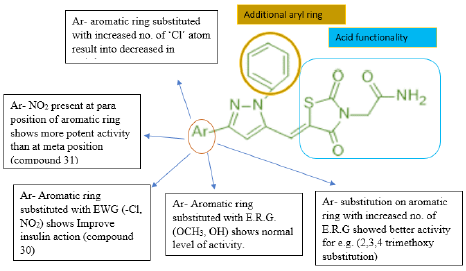
Mohd. Naim et. al prepared a new series of amide based TZD derivatives & screened them for their PPARγ agonist activity to exert anti-diabetic action. They performed molecular docking of all compounds, out of which compound 30 was found to be with best glide score & further selected for in vivo anti-diabetic activity on Streptococci induced diabetic rat model. Results are shown in (Table 17, 18) (figure 29 (a &b)) [41].
Structure-activity relationship:
(Figure 29) (Table 19,20)




Bio Isosteric Replacement for Thiazolidine 2,4 Diones:
The substitution of oxazolidine-2,4-dione, isoxazolidine- 3,5-dione, and succinimide rings can bioisosterically replace the electrophilic and hydrogen bonding-rich characteristics of the TZD nucleus Figure 27. With the aforementioned ring systems, the bio isosteric replacement of TZD typically results in the creation of potent and safe molecules with fewer adverse effects such as hepatotoxicity and cardiotoxicity, weight gain, fluid retention, and oedema, among others. (9) The oxazolidine 2,4-dione, Bioisosters of TZD, which exhibits equipotent anti-hyperglycemic activity as TZD derivatives, is produced when the oxygen atom replaces the sulfur atom in the TZD ring [42]. Succinimide Bioisosters of TZD. It has a similar five-membered ring structure to TZD’s succinimide Bioisosters, but the sulfur atom is replaced by methylene. This derivative sufficiently lowers plasma glucose levels [43]. In in vivo models, the TZD compound’s isoxazolidine-3,5-dione Bioisosters demonstrated excellent antihyperglycemic activity [44]. (Figure 30)


Conclusion
The TZD moiety has significant medicinal potential and plays a crucial role in the biological function of several essential molecules. The synthetic methods are straightforward and adaptable; As a result, new TZD analogs can be easily discovered. The researchers have the opportunity to create novel TZD molecules by modifying the third and fifth positions of the Thiazolidin-2,4-dione (TZD) scaffold with various moieties. TZDs are mostly used to treat diabetes but also have anti-inflammatory, anti-viral, cancerfighting, and antioxidants. This review article aimed to investigate the mechanism of action and pharmacological potential of TZDs for use as antimicrobial, antioxidant, and hypoglycemic agents. The general mechanism of actions & possible target sites for TZD are explained in this article, this information will surely benefit the researchers in developing a new drug candidate containing a TZD scaffold, as will those developing future drug molecules. This review’s findings suggest that TZDs are a promising class of drugs that can be used to create new, less toxic anti-diabetic, anti-fungal, anti-cancer, or antioxidant agents.


Acknowledgement’s
Authors are thankful to Principal, Government College of Pharmacy, Aurangabad for their constant support and encouragement. Authors also thankful to Dr. Manoj Damale sir, Associate Professor, Srinath College of Pharmacy, Aurangabad for their continuous support throughout the work.
References
- Kaur S (2017) Bioorganic Chemistry Synthetic and medicinal perspective of thiazolidinones: A review. Bioorg Chem 75: 406-423.
- Tahlan S, Verma PK, Sucheta (2017) Biological potential of thiazolidinedione derivatives of synthetic origin. Chem Cent J pp 11: 1-29.
- Naim MJ, Alam MJ, Ahmad S, Nawaz F, Shrivastava N, et al. (2017) Therapeutic journey of 2,4-thiazolidinediones as a versatile scaffold: An insight into structure activity relationship. Eur J Med Chem 129: 218-250.
- Tomasic T, Masic L (2009) Rhodanine as a Privileged Scaffold in Drug Discovery. Curr Med Chem 16(13): 1596-1629.
- Diekema DJ, Jones RN (2001) New drug classes Oxazolidinone antibiotics. Lancet 358(9297): 1975-1982.
- Kota BP, Huang TH, Roufogalis BD (2005) An overview on biological mechanisms of PPARs 51(2): 85-94.
- Jain VS, Vora DK, Ramaa CS (2013) Thiazolidine-2,4-diones: Progress towards multifarious applications. Bioorg Med Chem 21(7): 1599-1620.
- Parekh NM, Juddhawala KV, Rawal BM (2013) Antimicrobial activity of thiazolyl benzene sulfonamide-condensed 2,4-thiazolidinediones derivatives. Med Chem Res 22(6): 2737-2745.
- Chadha N, Bahia MS, Kaur M, Silakari O (2015) Thiazolidine-2,4-dione Derivatives: Programmed Chemical Weapons for Key Protein Targets of Various Pathological Conditions. Bioorg Med Chem 23(13): 2953-2974.
- Enchev V, Chorbadjiev S, Jordanov B (2002) Comparative study of the structure of rhodanine, isorhodanine, thiazolidine-2,4-dione, and thiorhodanine. Chemistry of Heterocyclic Compounds 38(9): 1268-1278.
- Nanjan MJ, Karvekar M, Suresh B, Prashantha Kumar B, Praveen T (2007) “Serum glucose and triglyceride lowering activity of some novel glitazones against dexamethasone-induced hyperlipidemia and insulin resistance. Indian J Pharmacol 39(6): 299.
- Srivastava T, Haq W, Katti S (2002) Carbodiimide mediated synthesis of 4-thiazolidinones by one-pot three-component condensation. Heterocyclic Compounds 58(38): 7619-7624.
- Romagnolo R, Baraldi PG, Salvador MK, Camacho ME, Balzarini J, et al. (2013) Anticancer activity of novel hybrid molecules containing 5-benzylidene thiazolidine-2,4-dione. Eur J Med Chem 63: 544-557.
- Li Q, Ayoubi AA, Guo T, Zheng H, Sarkar A, et al. (2009) Structure-activity relationship (SAR) studies of 3-(2-amino-ethyl)-5-(4-ethoxy-benzylidene)-thiazolidine-2,4-dione: Development of potential substrate-specific ERK1/2 inhibitors. Bioorg Med Chem 19(21): 6042-6046.
- Božić B, Rogan J, Poleti D, Rancic M, Trisovic N, et al. (2017) Synthesis, characterization, and biological activity of 2-(5-arylidene-2,4-dioxotetrahydrothiazole-3-yl) propanoic acid derivatives. Arab J Chem 10(Supp 2): 2637-2643.
- Ha YM, Park YJ, Kim JA, Park D, Park JY, et al (2012) Design and synthesis of 5-(substituted benzylidene) thiazolidine-2,4-dione derivatives as novel tyrosinase inhibitors. Eur J Med Chem 49: 245-252.
- Russell AJ, Westwood IM, Crawford MJH, Robinson J, Kawamura A, et al. (200) Selective small molecule inhibitors of the potential breast cancer marker, human arylamine N-acetyltransferase 1, and its murine homologue, mouse arylamine N-acetyltransferase 2. Bioorg Med Chem 17(2): 905-918.
- Salamone S, Colin C, Vuissoz IG, Kuntz S, Mazerbourg S, et al. (2012) Anti-proliferative activity in breast cancer cell lines and preliminary toxicological study. Eur J Med Chem 51: 206-215.
- Liu K, Guo TL, Hait NC, Allegood J, Parikh HI, et al. (2013) Biological Characterization of 3-(2-amino-ethyl)-5-[3-(4-butoxyl-phenyl)-propylidene]-thiazolidine-2,4-dione (K145) as a Selective Sphingosine Kinase-2 Inhibitor and Anticancer Agent. PLoS One 8(2): e56471.
- Sharma P, Reddy TS, Thummuri D, Senwar KR, Kumar NP, et al. (2016) Synthesis and biological evaluation of new benzimidazole-thiazolidinedione hybrids as potential cytotoxic and apoptosis-inducing agents. Eur J Med Chem 124: 608-621.
- El‐Adl K, El-Helby AGA, Sakr H, Ayyad RR, Mahdy HA, et al. (2021) Design, synthesis, molecular docking, anticancer evaluations, and in silico pharmacokinetic studies of novel 5‐[(4‐chloro/2,4‐dichloro) benzylidene] thiazolidine‐2,4‐dione derivatives as VEGFR‐2 inhibitors. Arch Pharm (Weinheim) 354(2): 2000279.
- El Adl K, Sakr H, Nasser M, Alswah M, Shoman FMA (2020) 5-(4-Methoxybenzylidene) thiazolidine-2,4-dione-derived VEGFR ‐ 2 inhibitors: Design, synthesis, molecular docking, and anticancer evaluations. Wiley 353(9): 1-16.
- Laxmi SV, Anil P, Rajitha G, Rao AJ, Crooks PA, et al. (2016) Synthesis of thiazolidine-2,4-dione derivatives: anticancer, antimicrobial and DNA cleavage studies. J Chem Biol 9(4): 97-106.
- Prabitha P, Justin A, Kumar TDA, Chinaswamy M, Kumar BRP (2021) Glitazones Activate PGC-1 α Signaling via PPAR- γ: A Promising Strategy for Ant parkinsonism Therapeutics. ACS Chem Neurosci 12(13): 2261-2272.
- Abdullah M, Ali M, Kour D, Kumar A, Bharate SB (2019) Discovery of Benzo [cd] indol-2-one and Benzylidene-thiazolidine- 2, 4-dione as New Classes of NLRP3 Inflammasome Inhibitors via ER-β Structure-Based Virtual Screening. Bioorg Chem 95: 103500.
- Elkamhawy A, Kim NY, Hassan AEH, Park JE, Yang JE, et al. (2019) Optimization study towards more potent thiazolidine-2, 4-dione IKK-β modulator: Synthesis, biological evaluation and in silico docking simulation. Bioorg Chem 92: 103261.
- Abdellatif KRA, Abdelgawad MA, Elshemy HAH, Alsayed SSR (2016) Design, synthesis, and biological screening of new 4-thiazolidinone derivatives with promising COX-2 selectivity, anti-inflammatory activity, and gastric safety profile. Bioorg Chem 64: 1-12.
- Mccarthy MW, Kontoyiannis DP, Cornely OA, Perfect JR, Walsh TJ (2017) Novel Agents and Drug Targets to Meet the Challenges of Resistant Fungi. 16(Supp 3): S474-S483.
- Marc G, Stana A, Pîrna A, Vlase L, Vodnar DC, et al. (2018) 3,5-Disubstituted Thiazolidine-2,4-Diones: Design, Microwave-Assisted Synthesis, Antifungal Activity, and ADMET Screening. SLAS Discov 23(8): 807-814.
- Marc G, Oniga SD, Vlase L, Chifiriuc MC, Pîrn A, et al. (2018) New N- (oxazolyl methyl)-thiazolidinedione Active against Candida albicans Biofilm: Potential Als Proteins Inhibitors. Molecules 23(10): 2522.
- Khare N, Kapoor A (2016) Antioxidant evaluation of 2, 4-thiazolidinedione and rhodanine derivatives 8(14): 38-46.
- Hossain SU, Bhattacharya S (2007) Synthesis of O-prenylated and O-geranylated derivatives of 5-benzylidene2, 4-thiazolidinediones and evaluation of their free radical scavenging activity as well as effect on some phase II antioxidant / detoxifying enzymes. Bioorg Med Chem Lett 17(5): 1149-1154.
- Kumar H, Deep A, Marwaha RK (2020) Design, synthesis, in silico studies and biological evaluation of 5-((E)-4-((E)-(substituted aryl/alkyl) methyl) benzylidene) thiazolidine-2,4-dione derivatives. BMC Chem 14(1): 25.
- Tahlan S, Verma PK, Sucheta (2018) Synthesis, SAR, and in vitro therapeutic potentials of thiazolidine ‑ 2, 4 ‑ Chem Cent J 12(1): 129.
- Marc G, Stana A, Oniga SD, Pîrna A, Valse L, et al. (2019) New Phenolic Derivatives of Thiazolidine-2,4-dionewith Antioxidant and Antiradical Properties. Molecules 24(11): 2060.
- Koppireddi S, Komsani JR, Avula S, Pombala S, Vasamsetti S, et al. (2013) Novel 2-(2,4-dioxo-1,3-thiazolidin-5-yl) acetamides as antioxidant and/or anti-inflammatory compounds. Eur J Med Chem 66: 305.
- https://www.who.int/health-topics/diabetes#tab=tab_1
- Chinthala Y, Domatti AK, Sarfaraz A, Singh SP, Arigari NK, et al. (2013) Synthesis, biological evaluation, and molecular modeling studies of some novel thiazolidinediones with triazole ring. Eur J Med Chem 70: 308-314.
- Bansal G, Singh S, Monga V, Thanikachalam PV, Chawla P (2019) Synthesis and biological evaluation of thiazolidine-2, 4-dione-pyrazole conjugates as antidiabetic, anti-inflammatory and antioxidant agents. Bioorg Chem 92: 103271.
- Datar PA, Aher SB (2012) Design and synthesis of novel thiazolidine-2, 4-diones as hypoglycemic agents. J SAUDI Chem Soc 20(Supp 1): S196-S201.
- Naim MJ, Alam MJ, Nawaz F, Naidu VG, Aaghaz S, et al. Synthesis, molecular docking, and anti-diabetic evaluation of 2, 4-thiazolidinedione based amide derivatives. Bioorg Chem 73: 24-36.
- Dow RL, Bechle BM, Chou T, Clark DA, Hulin B, et al. (1991) J = 1.4: 15381544.
- Saha S, New LS, Ho HK, Chui WK, Chun E, et al. (2010) Direct toxicity effects of sulfo-conjugated troglitazone on human hepatocytes. Toxicol Lett 195(2-3): 135-141.
- Christo A, Konstantinidou E, Jani C, Boussiotis VA (2020) The role of peroxisome proliferator-activated receptors (PPAR) in immune responses.















