




Method Development and Pharmacological study of Chemotherapeutic Agents
Krishnasarmapathy*
Hospital and Clinical Pharmacy, India
Submission: Juiy 14, 2017; Published: August 17, 2017
*Corresponding author: Krishnasarmapathy, Editoriai Board Member-Hospitai and Ciinicai Pharmacy, India, Tei: 9919188895; Email: drkrishnasarmapathy@yahoo.in
How to cite this article: Krishnasarmapathy. Method Development and Pharmacological study of Chemotherapeutic Agents. Open Acc J of Toxicol. 2017;1(4): 555568. DOI: 10.19080/OAJT.2017.01.555568
Abstract
The processes of method deveiopment for the pharmacoiogicai studies of chemotherapeutic agents (i.e., methoxyamine method deveiopment by using LC-MS/MS), were illustrated. To be more specific, a tetra-enzyme cocktail utilized for DNA adducts release was introduced. LC-MS/MS method for the analysis of methoxyamine modified DNA a basic sites incorporated in DNA was developed toward the DNA adducts released from DNA with the enzyme cocktaii. The methods were appiied to the drug effect and drug mechanism studies. The resuits in this work not oniy demonstrated the capabiiity of LC-MS/MS in soiving sophisticated pharmacoiogicai puzzies, but wiii provide usefui information guiding the preciinicai studies and ciinicai therapy deveiopment of the anti-cancer drugs. In the deveiopment of anti-cancer drugs, it is essentiai to study the pharmacological profiles of the drugs. Among the analytical tools utilized in the pharmacological studies, LC-MS/MS has gained increased popularity due to its unequivocal sensitivity and specificity, as well as the ability of handling a wide variety of compounds with relatively simple sampie preparation procedures. In this work, a brief review on the method rationai, instrumentations, anaiyticai method vaiidation, and work flow of the method development was included. Cancer is a worldwide problem. Finding novel compositions and methods for treating cancer is of interest. The treatment of cancer faiis into three generai categories: chemotherapy, radiation therapy and surgery. Often, therapies are combined since a combination of therapies increases the probabiiity the cancer wiii be eradicated as compared to treatment strategies utiiizing a singie therapy. Typicaiiy, the surgicai excision of iarge tumour masses is foiiowed by chemotherapy and/or radiation therapy
Pharmacological Studies of Anti-Cancer Drugs
Cancer is a woridwide probiem. Finding novei compositions and methods for treating cancer is of interest. The treatment of cancer faiis into three generai categories: chemotherapy, radiation therapy and surgery. Often, therapies are combined since a combination of therapies increases the probabiiity the cancer wiii be eradicated as compared to treatment strategies utiiizing a singie therapy. Typicaiiy, the surgicai excision of iarge tumour masses is foiiowed by chemotherapy and/or radiation therapy. Chemotherapeutic agents can work in a number of ways. For exampie, chemotherapeutics can work by interfering with ceii cycie progression or by generating DNA strand breaks [1]. If the cancer ceii is not abie to overcome the ceii cycie biockage or ceii injury caused by the therapeutic compound, the ceii wiii often die via apoptotic mechanisms. The use of a singie chemotherapeutic agent in the treatment of cancer, with or without surgery or radiation, has severai disadvantages. Commoniy, cancer ceiis deveiop resistance to the chemotherapeutic agent. Such resistance resuits either in the requirement for higher dosages of the drug and/or the renewed spread of the cancer. Chemotherapeutic agents can be toxic to the patient. Therefore, there is a practicai upper iimit to the amount that a patient can receive. However, if a second agent can be deveioped to inhibit the pathway causing resistance, cancer ceiis may become susceptibie to the effects of the chemotherapeutic agent [2]. The design of a drug to overcome resistance to the chemotherapeutic treatment of cancer shouid be approached with the goais of:
a. Finding a combination that reverses resistance and not mereiy improves the activity of the chemotherapeutic with respect to activity on the tumour, and
b. Finding a second drug that does not potentiate the toxic effects of the first chemotherapeutic agent
LCMS-MS
Detection and quantification-HPLC/MS/MS analysis
UDG-/- and UDG+/+ cells were exposed to pemetrexed (10µM) or 5-fluorouracil (10µM) for 6, 24, 48 and 72hrs. At indicated time points, ceiis were harvested and genomic DNA was extracted by phenol/chloroform. 40µg of DNA were incubated with 10 U of purified UDG (New England Biolabs) in 60µL of reaction buffer at 37 °C for 2hrs. The reaction products were dried at 35 °C in a Turbovap under a stream of nitrogen and reconstituted in 150µL 90% acetonitrile. The analyse was retained by an Atiantis Hiiis Siiica anaiyticai coiumn (2.1x100mm, 3.5µM) and eluted isocratically by a mixture of 90% acetonitrile and 10% 2.0mM ammonium format at a flow rate of 0.2ml/min. The detection was done by an API 3200MS/ MS mass spectrometer.
Material and Methods
Chemicals and solutions
MX•HCl, 2-deoxyribose, ribose, ammonium format, formic acid, isopropanoi, acetonitriie, BisTris, CT-DNA, O-(4- Nitrobenzyl) hydroxylamine, ethanol, sodium citrate, DNase I from bovine pancreas, NP1 from Peniciiiium citrinum, bovine intestinai ALP, and Tris EDTA buffer were obtained from Sigma- Aldrich (St. Louis, MO). Triethylamine, NaH2PO4, NaCl, ZnCl2, PBS, water saturated phenoi, and chioroform were purchased from Fisher Scientific (Fair Lawn, NJ) [3]. Tris base and 10% SDS were from Bio-Rad Laboratories (Hercules, CA). DMEM medium and L-glutamine were from Media tech (Manassas, VA). RNase and protease K were from Invitrogen (Carlsbad, CA). DNA 11- mers 5’-GCCGT-U-AGGTA-3’ and 5'-AGGTAGCCGT-U-3' were synthesized by Integrated DNA Technologies (Coralville, IA). Methanol was purchased from Pharmco- AAPER (Brookfield, CT). 1,1,1,3,3,3-Hexafluoro-2-propanol was obtained from Oakwood Products (West Columbia, SC) [4]. Uracil DNA glycosylase (with 10x enzyme buffer) was from New England Biolabs (Ipswich, MA) [5]. Acetic acid was from Mallinckrodt Baker (Phillipsburg, NJ). Hydrochloric acid was from EMD Chemicals (Gibbstown, NJ) [6]. Snake venom PDE I was obtained from Worthington Biochemical Corporation (Lakewood, NJ) [7]. Fetal bovine serum was purchased from Hyclone Laboratories (Logan, UT) [8]. TMZ was from Ochem (Des Plaines, IL) [9]. Deionised water was prepared by the Barnstead NANO pure® water purification system (Thermo Scientific, Waltham, MA, USA) [10].
Synthesis of MX-dR and MX-R
MX•HCl powder was dissoived in deionised water to a concentration of 1.0M. 2'-Deoxyribose and ribose powders were dissoived in deionised to make 1.0M soiutions, respectiveiy. Then each concentrated soiution was diiuted with deionised water to a concentration of 10mM [11]. To synthesize MX-dR, 10µL of MX•HCl solution (1.0M), 10µL of deoxy ribose solution (10mM) and 80µL of deionized water were pipette together into a 0.5mL micro centrifuge tube. Then the tube was kept in 70 for 2h. For the synthesis of MX-R, the IS, the 1 M MX•HCi soiution was diiuted with de-ionized water to 10mM. Then, 10µL of MX•HCl solution (10mM), 10µL of ribose solution (1.0M) and 80µL of de-ionized water were pipette together into a 0.5mL micro centrifuge tube [12]. Then the tube was kept in 70 again for 2h. Both reactions were stopped by 100xdiiution with deionised water. Then, the reaction products were kept in -4 tiii use.
LC-MS/MS and LC-MS instrumentations
The instrument system inciuded a Shimadzu HPLC system (Kyoto, Japan) composed of a solvent reservoir, a degasser (DGU- 20A3), a binary pump (LC-20AD) [13], a flow controller (CBM- 20A), and an auto sampler (SIL-20ACHT), together with an AB SCIEX 5500 QTRAP® 5500 mass spectrometer (Foster City, CA) controlled by Analyst software (version 1.5.1) [11].
LC-MS/MS of MX-dR and the IS
Chromatographic separation was carried out on a Thermo (West Palm Beach, FL) Hypercarb TM column (2.1x50mm, 5|iM) at ambient temperature (23) with a flow rate of 0.4mL/min. A two-soivent gradient, 5mM ammonium format [14] (NH4Fc, pH 3.5) (A)and 5mM NH4Fc (pH 3.5) in 67% methanol and 33% isopropanol (v/v) (B), was utilized for complete separation of MX-dR from the matrix interferences. At the beginning of the LC, 100% A was held for 1.0min. Then the content of A was dropped quickly from 100% to 40% within 1.0 to 1.1min. Next, the mobile phase was held at 40% A till 5.9min, followed by returning to 100% A at 6.0min. Before each run, there was an equilibration set as 5min. The coiumn eiuent was diverted to the waste before 2.49min, and then to the mass spectrometer between 2.50min and 3.10 min. At 3.11 min the flow was diverted to the waste again tiii the end of the run [15].
The mass spectrometer was operated at the positive-eiectro spray-ionization (ESI+) mode [16]. It was tuned by flow injection of a mixture of MX-dR (100ng/mL) and MX-R (100ng/mL) in the mobile phase (60% B) at a flow rate of 0.4mL/min. The source dependent parameters were as follows: curtain gas TM (CUR), 20; ion spray voltage (IS), 5500; temperature (TEM), 300; gas1 (G1), 60.0; gas 2 (G2), 60.0 [17]. The compound dependent parameters were as follows: De-clustering Potential (DP), 50.0; Entrance Potential (EP), 8.00. Detection of MX-dR and MX-R was based on MRM with the conditions set as foiiows: Coiiision Gas (CAD), low; Collision Energy (CE), 10.0; Collision Cell Exit Potential (CXP), 11.0; Dewell Time, 100ms [18]. Two MRM channeis: m/z 164 > 117, and m/z 180 > 102 were utiiized to monitor MX-dR and MX-R, respectiveiy.
LC-MS of the oligo nucleotides
The chromatography separation of the oiigonucieotides was carried out on an Xterra MSC18® column (2.0x50mm, 3.5|im, Waters, Milford, MA) by adjusting an existing method [19]. Isocratic separation was performed at ambient temperature (23) at a flow rate of 0.2mL/min with a mobile phase containing 86% 200mM 1,1,1,3,3,3- Hexafluoro-2-propanol [HFIP, adjusted to pH 7.0 with N,N,N-Tri ethylamine (TEA)] and 14% methanol (v/v). The column eluent was diverted to the waste before 2.00 min, and then to the mass spectrometer at 2.00min.
Negative-electro spray-ionization (ESI-) mode MS was operated with the source dependent parameters as the foiiowing: CUR, 20; IS, -4500; TEM, 400; G1, 40; G2, 40.The compound- dependent parameters were as foiiows: DP, -100; EP, -10. DNA 11-mers 5’-GCCGT-U-AGGTA-3’ and 5'-AGGTAGCCGT-U-3', 5'-GCCGT-AP-AGGTA-3', as well as 5’-GCCGT-(MX-AP)-AGGTA-3' were monitored with the Q1 Ml scan mode (selected reaction monitoring or SIR) in channel m/z 670.5 (M-5H), m/z 651.8 (M- 5H), and m/z 657.5 (M-5H), respectively. The Dewell Time was set as 100 ms for each channel. The DNA 11-mers were diluted with de-ionized water to 1µg/mL. For each analysis, 2µL of sample was injected onto the column.
Results and Discussion


Characterization of MX-dR and the IS with mass spectrometry Since MX-AP is bound to DNA strand, to realize the quantification, a tetra enzyme system was utilized to release MX-AP as MX-dR (Figure 1 & 2). By carrying out this enzymatic digestion, the quantification of MX-AP bound to DNA was converted to the quantification of the free small molecule, MX-dR. To achieve highly accurate and repeatable results, another small molecule, MX-R was synthesized as the IS [20]. After reaction, the two post-reaction mixtures were diluted with 5mM NH4Fc for 100 times, respectively, and infused into the mass spectrometer by a syringe pump at a flow rate of 5µL/min. As both MX-dR and the IS are more easily to form protonated species through ESI, ESI+ mode was utilized. As shown in Figure 3, MX-dR and the IS produced molecular ions at m/z 164 ((MX-dR+H)+) and m/z 180 ((MX-R+H)+), respectively. To achieve higher specificity in the quantification, the molecular ions were further dissociated with CID. From the resulted fragmentation pattern, two predominant fragments were observed at m/z 117 for MX-dR and m/z 102 for the IS, respectively. Therefore, the mass transition pairs m/z 164>117 for MX-dR and m/z 180>102 for the IS were utilized in the quantification work with MRM mode. Figure 2, Enzymatic release of MX-AP from the DNA backbone as MX-dR.

Digested DNA sample extraction
As the quantification was carried out toward the MX-dR in the enzyme digested DNA samples, several major interferences were expected: buffer salts (i.e., 4.69mMM BisTris, 145µM NaCl, and 14.1µM ZnCl2), proteins (i.e., protein impurities existing in the DNA samples and enzymes utilized in the digestion), and the dNs (with a total concentration of around 3mM). To avoid signal suppression and ion source contamination caused by these interferences, the analyse must be effectively separated from these interferences through on-line and/or off-line procedures. Two off-line extraction methods were tried in order to remove the matrix interferences.
A LLE method with a mixture of ethyl acetate and isopropanol (95:5, v/v) was tried. This method was effective in removing NaCl and ZnCl2. It was also able to remove over 90% BisTris. However, it failed to eliminate the dNs effectively. An SPE method with a caution exchange cartridge, the Oasis® MCX cartridge, was utilized under the intention of retaining all the dNs on the cartridge, yet collecting MX-dR from the cartridge pass-trough. To retain dA, dC, and dG, moderate acidic pH (i.e., pH 4) had to be utilized in sample loading. Under the same pH, however, dT was predominantly negatively charged, and could not be retained on the cartridge [21]. Besides, extra steps were still needed to separate the analyse from the buffer salts. As a result, the samples were simply processed by one-step acetonitrile precipitation to remove the proteins. Removal of the buffer salts and the dNs was left as a task in the LC method development.
LC separation of the analyse from the matrix interferences

Several columns (i.e., an YMC ODS-AQ® column, an Xterra MSC18® column, and a HypercarbTM column) were tried to obtain the separation between MX-dR and the other interference compounds. The YMC ODS-AQ® column was tried due to its capability of retaining highly polar compounds, and its compatibility with highly aqueous mobile phases. However, even when the percentage of the organic component (i.e., methanol) was dropped below 2%, no significant retention was observed for MX-dR. As MX-AP adduct can also be converted to MX-deoxy ribose 5'-phosphate (MX-dRp) in the enzymatic releasing of MX-AP through laminating ALP from the enzyme cocktail, ion-pairing chromatography with TEA was considered. In this test, the Xterra MSC18® column was utilized. By adjust the pH and the organic percentage of the mobile phase, MX- dRp could be retained on the column for up to 3 column volumes without causing significant tailing. However, all the 2'-deoxyribonucleotide mono phosphates (dNMPs) released after enzyme cutting could not be separated from the analyse effectively. In the work of Antonio et al. several sugar and sugar phosphates were separated on a porous graphitic carbon (PGC) column, the HypercarbTM column [22]. Because the graphite surface possesses a large amount of delocalized n-electrons, it is easy to induce electronic interaction with the analytes carrying polarisable or polarized groups [23]. And thus, the columns can provide strong retention to highly polar compounds. Another advantage of the PGC columns lies in their pH stability: they are stable throughout the pH rang as the structure of MX-dR is similar to those of the sugars, separation between the analyse and the matrix interferences was tried on this column. Isocratic elusion with a mixture of NH4Fc and organic solvents (i.e., methanol, acetonitrile, isopropanol, methanol/acetonitrile, or methanol/isopropanol) was able to retain MX-dR for at least 2 column volumes and achieve single symmetrical peak at the same time. Some of the conditions were also able to separate the analyse effectively from all the dNs and the inorganic salts. However, the separation between BisTris and MX-dR was always not enough due to the column's slightly retention to BisTris. Better separation between MXdR and BisTris can be achieved through utilizing lower percentage of weaker organic solvents, such as methanol, but the peak of MX-dR started to split. Besides, the retention times of the dNs were increased significantly (over 60min for dA and dG). Based on these reasons, a gradient elusion with the conditions described was finally adopted. With this LC method, MX-dR was able to be retained on the column for 2.8min, while all the dNs were eluted out after 3.2min (Figure 4). The BisTris was eluted out at 0.6 min. By applying the same LC condition, the IS was eluted out at 2.7min.
MX-AP DNA standard preparation
Since the quantification of MX-AP was realized through quantification of MX-dR released after enzyme digestion, DNA spiked with synthesized MX-dR would not be reliable in accurate quantification due to its invalidity in reflecting the digestion efficiency. Neither would the MX-AP DNA synthesized according to the protocol be reliable standards due to the uncertain amount of MX-AP sites it carries from batch to batch. Based on these considerations, a single strand DNA 11-mer with one MX- AP adduct located in the middle (i.e., 5'-GCCGT-(MX-AP)-AGGTA, the MX-oligo) was synthesized by modifying an existing protocol for AP-oligo synthesis [24]. Spiking the MX-oligo into blank CT- DNA resulted MX-AP DNA calibrators carrying all the necessary information required by the accurate quantification.

To monitor the synthesis process of MX-oligo, the starting material and product of each step of reaction were analyzed with the LC-MS method (Figure 5). As the remaining reactant was less than 1% of the original amount after each step of reaction, both steps were considered as complete. The U-mid- oligo was converted to equal amount of MX-oligo. To avoid high quantification background caused by the reaction between the excessive MX in the synthesis product and the AP sites generated through spontaneous hydrolysis during the enzyme digestion, the MX-oligo was purified on an Oasis® HLB SPE cartridge with a protocol adjusted from a published method [20]. By comparing the peak area of MX-oligo from the reaction product before and after purification, the recovery was determined as 94.2±1.6%.
As UDG is unable to remove uracil from the end of a DNA strand, UDG digestion on the U-end-oligo is not effective in producing AP-oligo (Figure 5). Further reaction with MX was not able to generate MX-oligo efficiently as well. Thus, the U-end- oligo was processed parallel with the U-mid-oligo as a negative control of the studies. In another word, the U-mid-oligo is able to reflect any background caused by excessive MX remaining in the purified oligomer products, or the background caused by MX-AP adducts formed from the reaction between MX and the AP sites generated spontaneously during the oligomer synthesis.
Method performance
To evaluate the performance of the developed methods in quantitative studies, a calibration curve was established with a linear calibration range of 0.358-22.7 MX-AP adducts/106 bases. The curve was weighted by the reciprocal of MX-AP concentration,1/x. The calibration equation has been shown in Table 1, and the linearity, represented by correlation coefficient R2, was 0.999±0.000. The accuracy and inter-assay precision of each point on the calibration curve ranged from 93.6-115% and 0.73-4.53%, respectively (Table 1).

The accuracy, intra-assay precision, and inter-assay precision of the analysis were determined through the quintuplicate calibrators at three concentration levels (low, medium, and high). All data were summarized in Table 2. The accuracy ranged from 86.7-98.2%; while the intra-and inter-assay precision varied from 1.02-5.25% and 3.14-3.78%, respectively. Here the accuracy was calculated by the relative deviation between a calculated concentration and the nominal concentration; while the precision was calculated by percent standard deviation.

Analysis of TMZ plus MX treated T98G cells in the cellular DNA analysis
In this section, the MX-AP concentration of every real sample was normalized with the method. The reason of carrying out this normalization procedure lies in its advantages in more accurate quantification. T98G cells treated with TMZ plus MX with different dosages and time spans were analyzed with the developed methods. A dose-effect profile and a time-effect profile were obtained afterward. From the dose-effect profile, a clear relationship between the dosage of TMZ plus MX and the concentration of MX-AP can be observed. For each dosage level of TMZ, the concentration of MX-AP was elevated with the increase of the MX dosage. Meanwhile, when the dosage of TMZ was increased, the profile of response was lifted systematically. These results are consistent with our hypothesis: higher concentration of MX blocks more AP sites; while higher concentration of TMZ generates more AP sites systematically. From the time-effect profile, when the treatment time increased from 6h to longer, the concentration of MX-AP decreased slightly at the beginning, and then reaches a relatively steady state after 24h treatment.
Analysis DNA samples from TMZ plus MX treated patient
The DNA samples from the patient with solid tumour enrolling in the phase I clinical trial of TMZ plus MX drug combination were analyzed. The time-response profile has been illustrated in. Determined from the profile, the concentration of MX-AP quickly reaches to the maximum after 4h treatment, and then decreased gradually below 0.500 MX-AP adducts/106 bases after 24h. The quick response of the patient to the treatment was consistent with our in vitro result. The clearance rate of MX-AP adducts, however, was much higher in the patient comparing to the cultured cells.
The reason for the difference is still under investigation. In fact, DNA samples from 4 patient enrolled in the phase I clinical studies were analyzed with our method, and only this one showed detectable signals for MX-AP. The PK profile of the patient indicated a significantly lower blood concentration of free MX. This may indicate extremely high MX incorporation into the patient's DNA. However, during the drug effect analysis, the amounts of MX-AP detected from the real samples were much lower comparing to the expectation. Under physiological conditions, DNA bases can be dissociated from the DNA strand through spontaneous hydrolysis at rate of 2 bases/106 bases in every 24h. Under the treatment of TMZ, although the amount of AP site generated after treatment has not been evaluated, it should be much higher than the AP sites generated by spontaneous hydrolysis. Assume that addition of MX will lead to 1 MX-AP adduct/106 bases after 24h treatment; at the same time, the white blood cell count in of the patient is 5x106 cells/ mL blood. As the blood drawn from each time point was around 5mL and the extracted DNA samples were typically dissolved in 15|iL of BisTris buffer before digestion, a calculation on the total amount of MX-AP adduct in the patient DNA samples can be roughly estimated as the following:
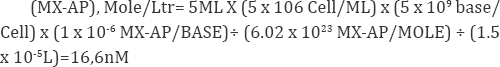
Since the total concentration of the dNs in a digested DNA sample is around 3.2mM, if convert the detection limit of MX-AP from standards to mole concentration:
(MX-AP), Mole/Ltr= (0.358 x 10-6 MX-AP/BASE) x 3.2Mm=1.15nM.
Based on the calculation, the MX-AP signal should be detectable in the patient DNA. However, due to the patients enrolled in the clinical trial were with solid tumour, the white blood cell count in these patient may be much lower than healthy donors. Meanwhile, as the cellular DNA was extracted in a relatively inefficient way, the recovery of the DNA may well below 100%. Last but not the least, the DNA extracted from the real samples can be easily contaminated with histon, SDS, and protease K [18]. All three kinds of impurities may greatly inhibit the releasing efficiency of the enzyme cocktail on the MX-AP adducts. In another word, before applying the developed method into the analysis of patient DNA samples, further optimizations must be carried out. The optimization can be done in two aspects. First, the DNA extraction and purification method can be further optimized. DNA extraction kit can be utilized for higher recovery and purity, as well as more reproducible results from sample to sample. Second, the enzyme digestion conditions can be further optimized. If necessary, different enzymatic system can be experimented.
Acknowledgement
The authors are thankful to TORRENT-Heuman pharma GMBH, CDRI Lucknow Pharmaceutical Industries Ltd, India for providing the standard and providing the research facilities
Conclusion
A tetra-enzyme cocktail containing DNA phase I, NP1, PDE I and ALP has been utilized for the quantitative releasing of MX- AP from the DNA backbone as MX-dR. A protein precipitation procedure was utilized in the sample preparation to remove protein interferences in the sample matrix. LC separation was realized on a HypercarbTM column for further separation of MX-dR and the IS from all the other interferences existing in the sample matrix. The MX-AP calibrators were prepared by spiking DNA 11-mers with one MX-AP site on each 11-mer into blank CT-DNA. A calibration curve ranged from 0.358-22.7 MX-AP adducts/106 bases were established, and the accuracy and precision were evaluated through a set of quality control calibrators. The feasibility of the method was tested by drug effect studies both in vitro (i.e., with the TMZ plus MX treated T98G cells)and in vivo (i.e., with the lymphocytes of the clinical patient treated with TMZ plus MX in a phase I clinical trial). The method we developed provided direct information on drug effect of MX, and thus will assist in providing dosimetric guidance in future clinical trials.
References
- Lawley PD, Phillips DH (1996) DNA adducts from chemotherapeutic agents. Mutat Res 355(1-2): 13-40.
- Neste EV, Cardoen S, Offner F, Bontemps F (2005) Old and new insights into the mechanisms of action of two nucleoside analogs active in lymphoid malignancies: fludarabine and cladribine (review). Int J Oncol 27(4): 1113-1124.
- Cardinal JW, Margison GP, Mynett KJ, Yates AP, Cameron DP, et al. (2001) Increased susceptibility to streptozotocin-induced beta-cell apoptosis and delayed autoimmune diabetes in alkylpurine-DNA-N-glycosylase- deficient mice. Mol Cell Biol 21(6): 5605-5613.
- Bulgar AD, Snell M, Donze JR, Kirkland EB, Li L, et al. (2010) Targeting base excision repair suggests a new therapeutic strategy of fludarabine for the treatment of chronic lymphocytic leukemia. Leukemia 24(10): 1795-1799.
- Seeberg E, Eide L, BjØrås M (1995) The base excision repair pathway. Trends Biochem Sci 20(10): 391-397.
- Middleton MR, Margison GP (2003) Improvement of chemotherapy efficacy by inactivation of a DNA-repair pathway. Lancet Oncol 4(1): 37-44.
- National Cancer Institute, Cancer Drug Information (2005) FDA Approval for Temozolomide.
- Newlands ES, Stevens MF, Wedge SR, Wheelhouse RT, Brock C (1997) Temozolomide: a review of its discovery, chemical properties, pre- clinical development and clinical trials. Cancer Treat Rev 23(1): 35-61.?
- Mitra G, Pauly GT, Kumar R, Pei GK, Hughes SH, et al. (1989) Molecular analysis of O6-substituted guanine-induced mutagenesis of ras oncogenes. Proc Natl Acad Sci USA 86(22): 8650-8654.
- Dianov G, Lindahl T (1994) Reconstitution of the DNA base excision- repair pathway. Curr Biol 4(12): 1069-1076.
- Wedge SR, Porteus JK, May BL, Newlands ES (1996) Potentiation of temozolomide and BCNU cytotoxicity by O(6)-benzylguanine: a comparative study in vitro. Br J Cancer 73(4): 482-490.
- Karran P, Macpherson P, Ceccotti S, Dogliotti E, Griffin S, et al. (1993) O6-methylguanine residues elicit DNA repair synthesis by human cell extracts. J Biol Chem 268(21): 15878-15886.
- Liuzzi M, Talpaert BM (1985) A new approach to the study of the base- excision repair pathway using methoxyamine. The Journal of Biological Chemistry 260: 5252-5258.
- Liu L, Taverna P, Whitacre CM, Chatterjee S, Gerson SL (1999) Pharmacologic Disruption of Base Excision Repair Sensitizes Mismatch Repair-deficient and -proficient Colon Cancer Cells to Methylating Agents Clin Cancer Res 5(10): 2908-2917.
- Liu L, Nakatsuru Y, Gerson SL (2002) Base excision repair as a therapeutic target in colon cancer. Clin Cancer Res 8(9): 2985-2991.
- Liu L, Yan L, Donze JR, Gerson SL (2003) Blockage of abasic site repair enhances antitumor efficacy of 1,3-bis-(2-chloroethyl)-1-nitrosourea in colon tumor xenografts. Mol Cancer Ther 2(10): 1061-1066.
- National Institute of Healt (2009) Methoxyamine and Temozolomide in Treating Patients with Advanced Solid Tumors.This work is licensed under Creative Commons Attribution 4.0 Licens DOI: 10.19080/QAJT.2017.01.555568
- Yang S, Liu L, Gerson SL, Xu Y (2003) Measurement of anti-cancer agent methoxyamine in plasma by tandem mass spectrometry with on-line sample extraction. J Chromatogr B Analyt Technol Biomed Life Sci 795(2): 295-307.
- Apffel A, Chakel JA, Fischer S, Lichtenwalter K, Hancock WS (1997) Analysis of Oligonucleotides by HPLC-Electrospray Ionization Mass Spectrometry. Anal Chem 67(9): 1320-1325.
- Gilar M, Bouvier ESP (2000) Purification of crude DNA oligonucleotides by solid-phase extraction and reversed-phase high-performance liquid chromatography. J Chromatogr A 890(1): 167-177.
- de Rock E, Taylor N (1977) An easy method of layering blood over Ficoll-Paque gradients. J Immunol Methods 17(3-4): 373-374.
- Antonio C, Larson T, Gilday A, Graham I, Bergstrom E, et al. (2007) Quantification of sugars and sugar phosphates in Arabidopsis thaliana tissues using porous graphitic carbon liquid chromatography- electrospray ionization mass spectrometry. J Chromatogr A 1172(2): 170178.
- Ross P, Knox JH (1997) Carbon-based packing materials for liquid chromatography: applications. Adv Chromatogr 37: 121-162.
- Roberts KP, Sobrino JA, Payton J, Mason LB, Turesky RJ (2006) Determination of apurinic/apyrimidinic lesions in DNA with high- performance liquid chromatography and tandem mass spectrometry. Chem Res Toxicol 19(2): 300-309.















