




Structure-Properties Relationship of Cu (II) – Paracetamol Based Complex. Density Functional Theory and Spectroscopic Studies
Hamrani O1, Zerrouk A1, Boutamine S1, Kellou-Tairi S2 and Hank Z1*
1USTHB - Laboratoire d’Electrochimie-Corrosion, Métallurgie et Chimie Minérale, Faculté de Chimie, BP 32 El Alia, Bab Ezzouar - Alger, Algérie
2USTHB - Laboratoire de Physico-Chimie Théorique et de Chimie Informatique, Faculté de Chimie, BP 32 El Alia, Bab Ezzouar - Alger, Algérie
Submission:October 23, 2020;Published: February 02, 2021
*Corresponding author: Hank Z, USTHB, Laboratoire d’Electrochimie-Corrosion, Métallurgie et Chimie Minérale, Faculté de Chimie, Alger, Algérie
How to cite this article: Hamrani O, Zerrouk A, Boutamine S, Kellou-Tairi S, Hank Z. Structure-Properties Relationship of Cu (II) – Paracetamol Based Complex. Density Functional Theory and Spectroscopic Studies. Nov Appro Drug Des Dev. 2021; 5(4): 555667. 5(4): 555667. DOI: 10.19080/NAPDD.2021.05.555668
Abstract
Our present work aimed to refine and optimize by a theoretical study the re-synthesize Cu (II) complex of N-acetyl-para-aminophenol (APAP) or paracetamol drug. Its structure structure-properties relationship is established. Although similar studies on synthesis and characterization have been conducted, the theoretical study has never been realized. Cu (II) complex of N-acetyl-para-aminophenol (APAP) drug has [Cu (APAP)2(OH)2.H2O] formula and noted Cu-APAP. It was characterized based on elemental analysis, conductivity, IR and thermal (TG/DTG), 1H NMR and electronic spectral studies. The value of molar conductance of the complex indicates a non-electrolyte nature.
The experimental results, IR, 1H NMR, 13C NMR and electronic spectra (UV) were compared to those obtained theoretically using the DFT method. 3D molecular modeling, the energetic, structural, and electronic parameters were carried out using density functional theory (DFT) at the B3LYP level for the ligand and UB3LYP for the Cu (II) complex, using Lacvp** as basis set.
Keywords: N-acetyl-para-aminophenol; Electronic parameters; Molecular modeling; Metal ions; Medicines; Kinetic properties
Abbreviations: APAP: N-acetyl-para-aminophenol; DFT: Density Functional Theory; TDDFT: Time-Dependent Density Functional Theory; PBF: Poisson Boltzmann finite element method
Introduction
From a chemical, biochemical, medicinal, and pharmacological point of view, the study of drug molecule complexation with various metal ions is an important field of research [1]. A great number of metal ions are required for the normal functioning of physiological processes. Furthermore, we ingest a certain number of metals such as medicines, dietetic factors, drinks; and live in close contact with a multitude of drugs. It is now well established that many pathological situations involve metal metabolism deregulation: therapeutic answers are thereby necessary. Even though the majority of medicines or compounds used in medicine are purely organic, the challenge is to improve the properties of these medicines by complexing them and investigating their interaction with trace elements present in the human organism. Complexation offers, indeed, the metal ion a multitude of coordination possibilities and a broad range of geometries. As a result of their thermodynamic and kinetic properties, and in certain cases their redox properties, metal complexes offer organic compounds new mechanisms of action which they usually lack, thus the importance of controlling all these properties [2-4].
Copper complexes have attracted a lot of attention because of their therapeutic applications as antimicrobials and antioxidants. We were interested in the complexation of this metal to the drug molecule N-acetyl-para-aminophenol (APAP). The aim of this study is to examine modification to the properties of an organic molecule that the metal can cause when it is coordinated to the former. Few studies have reported the complexation of metal ions by N-acetyl-para-aminophenol (APAP) [5-9]. The present study makes it possible to explain the reactivity of the complex which has proven to be a good antioxidant and a good anti-inflammatory agent [10].
N-acetyl-para-aminophenol (APAP) or paracetamol is an acylated aromatic amide, which was firstly introduced into medicine as an antipyretic/analgesic for home medication [11].
It was also known to be hepatotoxic in humans and various experimental animals at high concentrations [11-13]. Taking the presumed molecular mechanisms of analgesic activity as well as of the hepatotoxicity of APAP into consideration, there have been efforts to improve its analgesic activity while preventing its toxicity by modifying its structure [14].
The present work was built on the study of the interactions between N-acetyl-para-aminophenol (APAP) and Cu (II) metallic ion. DFT calculations carried out on both the structures of the ligand and the metal complex allowed us to investigate closer to the spectral features, and to propose the most fitting structures to the experimental data.
Experimental Section
a. Reagents and Chemicals
All chemicals used were analytical grade reagents without further purification. Copper (II) was used as chloride salt and was purchased from Fluka.N-acetyl-para-aminophenol (APAP: C8H9NO2 was provided by Algerian Pharmaceutical Industrial Group (SAIDAL).
b. Spectral and Physical Measurements
Elemental analyses (C, H, N and M) were performedat the Central Service of Analysis, CNRS (Solaize-Lyon, France). Infrared spectra (4000–400 cm-1) were recorded as KBr pellets on FTIR Nicolet Avatar 330 spectrophotometer. UV-vis absorption spectra were obtained in solid state or in DMSO solution from UV-Jasco V-650 spectrophotometer and recorded in 190-900 nm range. 1H and 13C NMR measurements were recorded with a RMN Bruker AM 300 spectrometer. Chemical shifts are expressed in ppm downfield from TMS. Thermogravimetric measurements (room temperature-600 °C) were recorded on a TA instrument Q500 thermogravimetric analyzer at a heating rate of 10 °C/min. Molar conductance were measured with HANNA instrument type HI 9932 microprocessors conductivity meter for the freshly prepared solutions in DMF or water. Melting points were determined in open glass capillaries on Stuart Scientific apparatus.
c. Synthesis of The Complex (Cu-APAP) The complex was prepared by mixing twice amount of N-acetyl-para-aminophenol (APAP) (18 mmol: 2.722 g) and 9mmol (2.180 g) of CuCl2 ·6H2O in MeOH/H2O (50/50, w/w) solvent. The reaction medium pH was adjusted to 9, using a concentrated ammonia solution. The mixture was then refluxed under backward flow at a temperature maintained under 60 °C for 6 hours and left to stand overnight. The precipitated complex was filtered, washed with distilled water followed by water- ethanol mixture (50:50 v/v proportions). The complex thus prepared, was dried in an oven for a few days at 60 °C.
Theoretical Calculations
Theoretical calculations for N-acetyl-para-aminophenol (APAP) and its Cu(II) complex were performed to optimize the geometry and to obtain the spectral features using Jaguar program included in the Schrodinger package for Linux platform, with the aid of Maestro GUI Schrödinger [15]. 3D molecular modeling of the ligand and the complex was carried out using density functional theory (DFT) at the B3LYP level for the ligand and UB3LYP for the Cu (II) complex, using Lacvp** as basis set [16]. The frequency calculations of the optimized geometries were done to confirm that the optimized structures achieved the minimum energy and to obtain the theoretical vibrational spectra by assuming C1 point group symmetry. All frequency calculations gave positive values. Vertical electronic excitations based on optimized geometries were computed using the time-dependent density functional theory (TDDFT) formalism in ethanol, using the Poisson Boltzmann finite element method (PBF).
Results and Discussion

The color, melting point, elemental analyses, and empirical formula of the prepared complex noted Cu-APAP, are given in Table 1. A good confirmation of the complex molecular weight was obtained from its mass spectrum which shows a molecular ion peak at m/z = 416 Figure 1) (calc. = 417.99). The complex is stable to air and moderately soluble in water and most organic solvents. The molar conductance value measured at 20 °C is in the range of no electrolytes [17].

d. IR Spectra
The ligand coordination sites involved in bonding with the metal ion have been determined by careful comparison of the infrared absorption spectrum of the complex with that of the free ligand (Figure 2). The vibrational assignments were carried out using density functional theory (DFT) at the B3LYP level for the ligand and UB3LYP for the Cu (II) complex. The experimental and theoretical FT-IR spectra of Cu-APAP are given in Figure 3, while the corresponding frequencies along with the assignments are gathered in Table 2.


The most interesting feature in the infrared spectra of this complex is the shift and decreasing in the intensities of bands 3200-3100 cm-1 region. The subsequent ligation of APAP through phenolic hydroxyl group oxygen atom seems a plausible explanation. The widening of the vibration band indicates the involvement of the OH group in an intra- or intermolecular hydrogen bond without deprotonation. The bands in the range 3400-3500 cm-1 can be assigned to NH which is not shifted; we can conclude that the NH group is not involved in the coordination process. The band at 1656 cm-1, assigned to the amidic C=O vibration, is shifted by comparison to its frequency in the free APAP (1666 cm-1); we can conclude that the carboxylic group is probably involved in a strong intermolecular hydrogen bond. Band at 1281 cm-1in the free ligand and attributed to C-N vibration is not changed in the complex (1279 cm-1); that confirms one more time that the NH group is not involved in coordination. The free ligand has a band in the C-O region, namely at 1228 cm-1. On complexation the maxima are significantly shifted to lower frequencies (1210 cm-1) with a marked change of intensity. This situation confirms that the C-O group is probably involved in coordination.
Complementary evidence of the bonding is also shown by the fact that new bands of medium intensity in the IR spectrum of the complex appear at 500-480 cm-1 and 480-450 cm-1 assigned to μ(Cu-Ophenolic) and μ(Cu-OH) stretching vibrations. The corresponding theoretical calculated modes were found in the regions 480-450 cm-1 and 440-400 cm-1, respectively.
The comparison of the experimental and the calculated spectral data, presented in Table 2, shows a good agreement with small differences. The difference that appears between those two spectra, illustrated in Figure 3, can be attributed to a fact that the experimental spectrum is obtained from a solid complex, in which interactions within the crystal system exist, but the calculated one results from an optimization in vacuum [18].

A correlation graph between the calculated and experimental wavenumbers for APAP and Cu-APAP complex, illustrated in Figure 4, highlights the linear relationship with correlation coefficients (R2) values = 0.9974 and 0.9983 for APAP and Cu-APAP complex, respectively. The respective equations are: νexper. = 0.9927 vcalc. + 13.895 and νexper. = 1.0564 vcalc. – 62.929.

e. 1H NMR Spectra
NMR spectroscopy is currently used for the determination of the structure of organic molecules or organic-based compounds. The 1H NMR spectra of APAP and its complex are shown in Figure 5. The experimental spectrum (Figure 5a) is obtained by dissolution of APAP in deuterated dimethylsulfoxid DMSO d6. The 1HNMR spectrum displays five signals in 1:1:2:2:3 ratios. The signal (1H) at δ = 9.66 ppm is due to the amidic group. The second one observed at 9.14 ppm (1H) is assigned to the proton of the phenolic OH resonance. The two signals located at 7.35 and 6.69 ppm and each corresponding to the resonance of two protons are attributed to the benzene ring protons. The last one at 2.07 ppm describes perfectly the CH3 protons resonances.

The 1H NMR spectrum of the complex (Figure 5b) presents the same signals when it is compared to the free APAP one. In fact, the signal of NH proton appears at 9.68 ppm corresponding to the same value recorded in unbound APAP. The OH protons resonances appear between 9.00 and 9.12 ppm. This very slight shift proves that the OH function is involved in the coordination process. The benzene protons are related by the singlet at 7.32 ppm (4H) and the doublets located, respectively at 6.78 ppm (2 H) and 6.66 ppm (2H). Finally, the signal at 1.97 ppm is due to the methyl protons (6H).
The new signal at 3.35 ppm could correspond to the overlap of the protons’ resonances of water molecules and those of the bound hydroxyl groups. The last signal at 2.5 ppm is due to residual nondeprotonated DMSO molecules.
f. 13C NMR Spectra
The results of this technique commonly used in the identification of organic molecules, correlated with proton NMR analyses, have contributed to the rigorous identification of the ligand, proving once again, the purity of the compound used (Figure 6).

Considering the two electron-attracting effects, it is fast to allocate the signals present in the APAP 13C spectrum (Figure 6a). The attractor carbon of carbonyl resonates around 171 ppm. The attractive inductive effect of this group and its conjugation with the free doublet of nitrogen of NH group is also exerted on the benzene ring carbons, which resonate in a ratio of 1:1:2:2 at 155.39, 131.69, 123.38 and 116.20 ppm. These chemical shifts indicate a significant deviation from the standard value of 128.5 ppm. The last carbon, that of the methyl group, influenced by the attraction of the C=O group shows a resonance at 23.51 ppm. The spectrum of the complex displays a similar spectrum suggesting that the APAP molecule is bound to the cupric ion without any alteration. This study corroborates once again the proposed structure for the copper complex.
g. Electronic Spectra
Vertical electronic excitations based on optimized geometries were performed using the time-dependent density functional theory (TDDFT) formalism. The UV-visible spectra are recorded in DMSO solution at 298 K.
The spectrum of APAP presented in Figure 7a displays one band centered at 40000 cm-1 and a shoulder at 37714 cm-1. The first band can be attributed to π −π * transition within the phenolic ring, while the second band could be due to n-π* transition within -C=O group. This bad resolution can be explained by the overlapping of the two transitions. The calculated electronic spectrum (Figure 7b) shows two bands at 62500 cm-1 and 38461 cm-1. Upon complexation, these transitions shift to longer wavelengths.

The electronic spectrum of Cu-APAP complex shown in Figure
8a, displays three bands. The former is sharp at 45454 cm-1 while
the second is a shoulder centered at 33333 cm-1. The two bands
are assigned, respectively to n→π *of phenolic group and
n→π *transitions of carbonyl group. The last band is broad and
centered around 20000 cm-1. It corresponds to the overlapping of
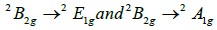 d-d transitions of Cu (II) ion
in a square plane geometry with a CuO4 environment [19-24].
On comparison with the theoretical spectrum (Figure 8b), a
good agreement is observed since almost the bands were at more
or less the same positions and with similar pattern. This confirms
the square planar geometry of the Cu (II) complex.
d-d transitions of Cu (II) ion
in a square plane geometry with a CuO4 environment [19-24].
On comparison with the theoretical spectrum (Figure 8b), a
good agreement is observed since almost the bands were at more
or less the same positions and with similar pattern. This confirms
the square planar geometry of the Cu (II) complex.

h. Thermal Analysis
Heating rates were controlled to 10 °C/min under a nitrogen atmosphere and weight loss was measured from room temperature to 600 °C for the studied complex. The two curves TG/DTG for APAP (Figure 9a) present only one weight loss stage on a 220-230 °C temperature range. Decomposition stages, in the studied temperature range (Figure 9b), the elimination product as well as found and calculated percentages weight loss of Cu-APAP of formula; [Cu(C8H9NO2)2(OH)2]. H2O (417.99 g/mmol) are given in Table 3.


The TG curve indicates a significant stability of the complex up to T= 120 °C. Above this temperature, the complex starts to decompose and the thermogram presents three main distinct stages maximized at 210, 300, and 400 °C on the DTG curve.
According to the hypothesis that the final residue is CuO, the recorded total loss would be appropriate for the computed value based on the suggested structural formulation of the complex. The small discrepancy between calculated and measured values may be due to incomplete decomposition of the substance at 600 °C.
Theoretical Study
i. Geometric Structures
We report here the main theoretical results as illustrations made with the GUI Maestro Schrodinger [25]. A comparison between the optimal geometric parameters (bond lengths, bond angles and torsion angle) of the free ligand and the ligand complexed with copper metal is investigated in Figure 10. The length of C-O, is found to be higher in complex than in the free ligand, revealing its involvement in bonding with metal and indicating strong ability of copper ion to attract oxygen atom. We give below geometric parameters around metal in the optimized complex.
The analysis of the optimized geometric parameters related to the atoms involved in the coordination shows that the geometry of the complex suggests a non-regular square planar structure.
j. Electronic Properties
The electronic structure of the studied compounds can be investigated by the quantum chemical parameters like energy of the highest occupied molecular orbital (EHOMO), energy of the lowest unoccupied molecular orbital (ELUMO), LUMO-HOMO energy gap (ΔE) and natural atomic charges calculated by natural population analysis. The finding data are grouped in Table 4 and illustrated in Figures 11 and 12. The HOMO and the LUMO are important parameters in organometallic chemistry especially in chemical reactivity, also called the frontier molecular orbital. ΔE is an important stability index helping to characterize the chemical reactivity and kinetic stability of the compound [26].
It is important to note that these theoretical parameters provide information about the reactive behavior of the investigated compounds. The analysis of natural atomic charges shows a change in values between the free ligand and the complexed one (Figure 11).
The analysis of the spatial distribution of the frontier molecular orbitals indicates that in the free APAP, the HOMO is localized over more atoms of APAP, whereas the LUMO is mainly localized on the phenolic hydroxyl group, NH and C=O groups of APAP (Figure 12).


In the Cu complex, the HOMO is distributed on the ligand and especially on the carbon atoms of the cycle (Figure 12). The LUMO is mainly localized on the metal and the oxygen atom of ligand molecule and also overall the atoms surrounding the ligand. Figure 12 shows that the central ion Cu (II) brings an important contribution to the LUMO, which suggests electrophilic attack sites. Frontier orbital’s energies of APAP and its metal complex are gathered in Table 4.
All the energies of the frontier molecular orbitals, including those of spin-orbitals, are negative, which indicates the stability of the compounds [27]. The prepared metal complex has the smallest energy gap, which suggests a lower kinetic stability than N-acetyl-para-aminophenol (APAP); it is the most reactive since it easily offers electrons to an acceptor [28].


Conclusion
In this study, we have reviewed the reactivity of N-acetylpara- aminophenol (APAP) towards copper. Copper complex has been well characterized by 1H NMR, 13C NMR, IR spectroscopy and thermal analysis. The IR spectral data suggests that APAP behaves as a monodentate coordinated ligand to the metal ions via the phenolic oxygen. All the spectral data corroborate perfectly our structural assumptions.
From this study, we found that the DFT/B3LYP/ Lacvp** method gives satisfactory results for this type of complex. Results obtained from DFT calculations corroborate perfectly the experimental results concerning complex stability and its IR and UV-visible spectra. Indeed, a very good agreement is observed when the experimental spectroscopic data are compared to the theoretical ones. We have also confirmed that Cu-APAP complex has a distorted square planar geometry. All the energies of the frontier molecular orbitals, including those of spin-orbitals, are negative, which indicates the stability of the compound. The experimental results concerning the reactivity of the generated complex were prognosticated by DFT calculations. In fact, the frontier molecular orbital energy gap reveals that the Cu complex was predicted to be more reactive than the free ligand.
Acknowledgements
The authors thank DGRSDT (La Direction Générale de la Recherche Scientifique et du Développement Technologique - MESRS) of Algeria for financial support.
References
- Moamen SR, Sharshar T, Khaled ME,Zein KH (2013) Physicochemical impact studies of gamma rays on “aspirin” analgesics drug and its metal complexes in solid form: Synthesis, spectroscopic and biological assessment of Ca(II), Mg(II), Sr(II) and Ba(II) aspirinate complexes.Journal of Molecular Structure 1047: 37-47.
- Anilanmert B (2012) Therapeutic Organometallic Compounds, Pharmacology, Available: Luca Gallelli, IntechOpen.
- Hartinger CG, Dyson PJ (2008) Bioorganometallic chemistry-from teaching paradigms to medicinal applications. Soc. Rev. 38: 391-401.
- Haas KL, Franz K (2009) Application of metal coordination chemistry to explore and manipulate cell biology.JChem Rev 109: 4921-4960.
- Lawal A, Obaleye JA (2007) Synthesis, characterization and antibacterial activity of aspirin and paracetamol metal complexes.Biokemistri 19: 9.
- El-Megharbel SM, Reham ZH, Moamen SR (2014) Preparation, spectroscopic, thermal, antihepatotoxicity, hematological parameters and liver antioxidant capacity characterizations of Cd(II), Hg(II), and Pb(II) mononuclear complexes of paracetamol anti-inflammatory anti-inflammatory drug.Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 131:534-544.
- Ionut L, Georgeta S, Gabriela V, Germaine S, et al. (2013) Synthesis and solid-state characterization of Zn(II) metal complex with acetaminophen. Rev Chim(Bucharest) 64: 1127.
- Sangita PK, Farhad MA, Sania S, Zakir MdS, Mohammad AH et al. (2012) Study of Differential Scanning Calorimetry of complex of Magnesium Sulfate with Aspirin, Paracetamol and Naproxen. Bangladesh Pharmaceutical Journal 15(1): 7-12.
- S El-Shahawy, Ahmed SM, SayedNKh (2007) INDO/SCF-CI calculations and structural spectroscopic studies of some complexes of 4-hydroxyacetanilide. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy66, 143-152.
- Espinosa Bosch MAJ, Ruiz Sanchez F, Sanchez Rojas C, Bosch O (2006) Determination of paracetamol: Historical evolution.Journal of Pharmaceutical and Biomedical Analysis 42(3): 291-321.
- Jollow DJ, Mitchell JR, Potter WZ, Davis DC, Gillette JR, (1973) Acetaminophen-Induced Hepatic Necrosis. II. Role of Covalent Binding In Vivo. JPharmacol Exp Ther 187(1): 195-202.
- Prescott LF (1983)Paracetamol Overdosage. Drugs 25: 290-314.
- Vermeulen NPE, Bessems JGM, Van de Straat R (1992)Molecular Aspects of Paracetamol-Induced Hepatotoxicity and its Mechanism-Based Prevention. Drug Metab Rev24: 367-407.
- El-Megharbel SM, Reham ZH, Moamen SR (2014)Preparation, spectroscopic, thermal, antihepatotoxicity, hematological parameters and liver antioxidant capacity characterizations of Cd (II), Hg (II), and Pb (II) mononuclear complexes of paracetamol anti-inflammatory drug. Spectrochimica Acta Part A:Molecular and Biomolecular Spectroscopy 131: 534-544.
- Jaguar, version 8.2, Schrödinger LLC, 2013, New York, NY.
- Bochevarov D, Harder E, Hughes TF, Greenwood JR, Braden DA, et al. (2013) Jaguar: A high‐performance quantum chemistry software program with strengths in life and materials sciences. Int J Quantum Chem 113: 2110-2142.
- Geary WJ (1971) The use of conductivity measurements in organic solvents for the characterization of coordination compoundsCoordination Chemistry Reviews 7: 81-122.
- Orif MI, Abdel-Rhman MH (2015)Synthesis, spectral and structural studies on some new isonicotinicthiosemicarbazide complexes and its biological activity. Polyhedron 98: 162-179.
- Hassaneien MM, Gabr IM, Abdel-Rhman MH, El-Asmy AA (2008)Synthesis and structural investigation of mono- and polynuclear copper complexes of 4-ethyl-1-(pyridin-2-yl) thiosemicarbazide. Spectrochim. ActaPart A Mol BiomolSpectrosc 71: 73-79.
- Silverstein R, Webster F (2006) Spectrometric identification of organic compounds. John Wiley & Sons, New York.
- Mostafa MM (2007) Spectroscopic studies of some thiosemicarbazide compounds derived from Girard's T and PSpectrochim ActaPart A: Mol BiomolSpectrosc66(2): 480-486.
- K Nakamoto (1978) Infrared and Raman spectra of inorganic and coordination compounds. Wiley Online Library.
- Richard L Carlin (1969) Inorganic Electronic Spectroscopy /Ed. A. B. P. Lever. Elsevier, Amsterdam.
- Kellett, Howe O, O’Connor M, McCann M, Creaven BS, et al. (2012) Radical-induced DNA damage by cytotoxic square-planar copper (II) complexes incorporating o-phthalate and 1,10-phenanthroline or 2,2′-dipyridyl. Free Radical Biology and Medicine, 53: 564-576.
- (2013)Maestro, version 9.6, Schrödinger LLC, New York, NY.
- El-Gammal OA, Rakha TH, Metwally HM, Abu El-Reach GM (2014) Synthesis, characterization, DFT and biological studies of isatinpicolinohydrazone and its Zn (II), Cd (II) and Hg (II) complexes.Spectrochim Acta A 127: 144-156.
- Dede B, Ozmen I, Karipcin F (2008) Synthesis, characterization, catalase functions and DNA cleavage studies of new homo and heteronuclear Schiff base copper (II) complexes.Polyhedron 28: 3967-3974.
- Hamrani O, Boutamine S, Tairi-Kellou S, Hank Z (2017)Copper-Drug Based Complexes: Antimicrobial, Antioxidant and Pharmacological Study. Global Journal of Nanomedecine 3(2): 555610.















