




Development and Invitro Characterization of Probenecid Rapidly Disintegrating Tablets by Using Different Co-Processed Super Disintegrants
Amareshwar S*1, Vishwanadham Y2, Prashanthi Y3 and Sreekanth D4
1St.Mary’s Pharmacy College Deshmukhi, India
2Vishnu Institute of Pharmaceutical Education and Research, India
3Nizam Institute of Pharmacy, Deshmukhi, India
4Palamuru University University, India
Submission: November 21, 2017; Published: December 18, 2017
*Correspondence author: Amareshwar S, St.Mary's Pharmacy College Deshmukhi, Pochampally, Yadadri- Bhongir, Telangana, Email: amar.pharma99@gmail.com
How to cite this article: Amareshwar S, Vishwanadham Y, Prashanthi Y, Sreekanth D. Development and Invitro Characterization of Probenecid Rapidly Disintegrating Tablets by Using Different Co-Processed Super Disintegrants.Nov Appro Drug Des Dev. 2017; 3(3): 555611.DOI:10.19080/NAPDD.2017.03.555611
Abstract
In the present work, an attempt has been made to develop fast disintegrating tablets of Probencid. Novel method of co processed super disintegrates technology was employed to formulate the tablets. All the formulations were prepared by direct compression method. The blend of all the formulations showed good flow properties such as angle of repose, bulk density, tapped density. The prepared tablets were shown good post compression parameters and they passed all the quality control evaluation parameters as per I.P limits. Among all the formulations F4 formulation showed maximum drug release i.e, 98.16% in 30min hence it is considered as optimized formulation. The F4 formulation contains CP2 as super disintegrate in the concentration of 50mg CP 2 contains CCS and CP in 1:2ratio.
Keywords: Probenecid; Co processed super disintegrates CCS; CP
Introduction
Over the past three decades, orally disintegrating tablets (ODTs) have gained much attention as a preferred alternative to conventional oral dosage forms such as tablets and capsules. An ODT could be a solid dose type that disintegrates and dissolves within the mouth either on or below the tongue or within the buccal cavity. Solid oral dose forms, particularly tablets, stay one amongst the foremost widespread due to benefits like patient convenience, easy storage and dispensing, dose accuracy and simple manufacturability. Major challenge for tablets producing comes from the flow properties of the materials to be compressed [1-3]. Most of the formulations & gt; 70% contain excipients at higher concentration than active drug. In recent years drug formulation scientists have recognized that single-component excipients don't invariably give the requisite performance to permit bound active pharmaceutical ingredients to be developed or manufactured adequately. Hence, there's a need to possess excipients with multiple characteristics designed into them like better flow, low/no moisture sensitivity, superior compressibility and fast disintegration ability. Excipients with improved practicality will be obtained by developing new chemical excipients, new grade of existing materials and new combination of existing materials. New mixtures of existing excipients are an interesting possibility for up excipients functionality as a result of all formulations contains multiple excipients. One such approach for up the practicality of excipients is co-processing of 2 or additional excipients. Comparison of ODTs and their typical various over as they disagree within the pharmacokinetic profile and bioavailability of a similar dose of drug (Table 1).

Causative relationship for mentioned variations could also be deemed to the drug chemistry property, formulation structure, producing method or all of them. Pharmacokinetic profile variations by suggests that of upper drug plasma levels and general exposure may be deemed to the pregastric absorption that permits the avoidance of first-pass metabolism, so result the security and effectiveness of the drug. the best characteristics of a drug allow dissolution within the mouth associate degreed pregastric absorption from an ODT includes having no bitter style, dose as low as potential little to moderate relative molecular mass, good solubility in water and saliva, partially non-ionized property at the oral pH. Conversely, having short half-life and wish for frequent dosing, heavily bitter or unsuitable style while not presumably of masking, demand of changed release may incapacitate a drug for ODTs. The quick dissolving property of the ODTs needs fast ingress of water into pill matrix so needs some basic approaches like maximizing the porous structure of the tablet, incorporation of appropriate disintegrating agent and use of extremely soluble excipients within the formulation Figures 1 & 2.


Excipients used in ODTs contain a minimum of one super disintegrants having mechanism of wicking, swelling or both, diluents, a lubricator and optionally a swelling agent, a permeabilizing agent, sweeteners and flavourings a part of the method of crucial if a product is an ODT involves testing a product to see however long it takes to disintegrate. Determination of disintegration time seems to be technique dependent [4-6]. Some methods are additional discriminating than others. to supply each a typical for and consistency in disintegration testing, we have a tendency to advocate that candidates use the USP method for disintegration testing. Three but, alternative methods that may be related to with or demonstrated to supply results equivalent to the USP method can also be used and submitted to determine disintegration time.
Drug Profile
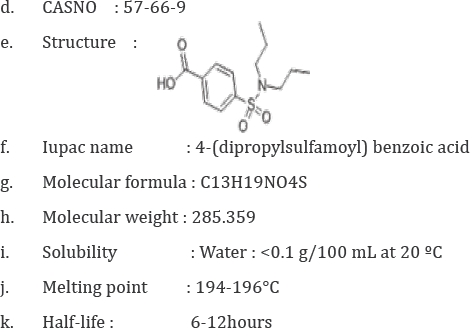
a. Drug name:Probenecid
b. Synonyms: 4-((Dipropylamino)sulfonyl) benzoic acid, Probenecid acid, 4-(N,N- Dipropylsulfamoyl) benzoesaeure
c. Description : The prototypical uricosuric agent. It inhibits the renal excretion of organic anions and reduces tubular reabsorption of urate. Probenecid has also been used to treat patients with renal impairment, and, because it reduces the renal tubular excretion of other drugs, has been used as an adjunct to antibacterial therapy.
Methodology
Materials
Probenecid, Microcrystalline cellulose, Cross carmellose sodium, Cross povidone, Magnesium stearate, Talc.
Equipments used
Weighing Balance, Tablet Compression Machine (Multistation), Hardness tester, Vernier callipers, Roche Friabilator, Dissolution Apparatus, UV-Visible Spectrophotometer, pH meter, FT-IR [7-9].
Preformulation studies
The goals of the preformulation study are:
a. To establish the necessary characteristics of a new drug substance.
b. To determine its kinetic release rate profile.
c. To establish its compatibility with different excipients.
Hence, preformulation studies on the obtained sample of drug include colour, taste, solubility analysis, melting point determination and compatibility studies and flow properties.
Determination of Absorption Maximum (λmax):
Absorption maximum is the wavelength at which maximum absorption takes place. For accurate analytical work, it is important to determine the absorption maxima of the substance. Probenecid was weighed accurately 10mg and transferred to 100ml volumetric flask, dissolved in 6.8pH phosphte buffer and the final volume was made up to 100ml with 6.8pH phosphte buffer to get a stock solution (100μg/ml). From the stock solution, 1ml was pipette out in 10ml volumetric flask and the final volume was made up to 10ml with 6.8 pH phosphte buffer to get 10μg/ml. Then this solution was scanned at 200-400nm in UV-Visible double beam spectrophotometer UV-3200, Labindia, India to get the absorption maximum (λmax) Figure 3.

Construction of Probenecid calibration curve with phosphate buffer PH 6.8
100mg of Probenecid was dissolved in 100ml of 6.8pH phosphte buffer to give a concentration of 1mg/ml (1000μgm/ ml). From the above standard solution (1000μgm/ml) 1ml was taken and diluted to 100ml with.1N HCL to give a concentration of 0.01mg/ml (10μgm/ml). From this stock solution aliquots of 0.5, 1, 1.5,2, 2.5ml were pipette out in 10ml volumetric flask and the volume was made up to the mark with )6.8pH phosphate buffer to produce concentration of 5,10,1,20 and 25μgm/ml respectively. The absorbance (abs) of each conc. was measured at respective (λmax) i.e., 259nm.
Drug- excipient compatibility studies by FT-IR
The compatibility between the pure drug and excipients was detected by FTIR spectra obtained on Bruker FTIR Germany(Alpha T).The potassium bromide pellets were prepared on KBr press by grounding the solid powder sample with 100 times the quantity of KBr in a mortar. The finely grounded powder was then introduced into a stainless steel die and was compressed between polished steel anvils at a pressure of about 8t/in2. The spectra were recorded over the wave number of 8000 to 400cm-1. Flow properties: Angle of Repose, Loose bulk Density, Tapped bulk density, Carr's consolidation index, Hauser’s ratio.
Preparation of tablets
Composition of Probenecid Dispersible Tablet by direct compression is shown in Table 2 all the ingredients were weighed. Required quantity of drug and excipient mixed thoroughly in a polybag. The blend is compressed using rotary tablet machine-10 station with 9mm flat punch, B tooling. Each tablet contains 250mg Probenecid and other pharmaceutical ingredients [10-12].

Post Compression Parameters
Evaluation of uncoated tablets
Shape and color: The tablets were examined under a lens for the shape of the tablet and colour by keeping the tablets in light.
Uniformity of thickness: Randomly 10 tablets were taken from formulation batch and their thickness (mm) was measured using a Vernier calipers.
Hardness test: The hardness of the tablets was determined using Monsanto hardness tester. It is expressed in Kg/cm2. Six tablets were randomly picked from each formulation.
Friability test: It is the phenomenon whereby tablet surfaces are damaged and show evidence of lamination or breakage when subjected to mechanical shock or attrition. The friability of tablets was determined by using Roche Friabilator (Lab India, FT 1020). It is expressed in percentage (%). Ten tablets were initially weighed [W (initial)] and transferred into Friabilator. The Friabilator was operated at 25 rpm for 4 min or run up to 100 revolutions. The tablets were weighed again [W (final)]. The How to cite this article: Amareshwar S, Vishwanadham Y, Prashanthi Y, Sreekanth D. Development and Invitro Characterization of Probenecid percentage friability was then calculated by
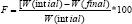
Weight variation test: The tablets were selected randomly from each formulation and weighed individually to check for weight variation. The U.S Pharmacopoeia allows a little variation in the weight of a tablet. The % deviation in weight variation is shown in table.
Drug Content estimation
The content uniformity test is used to ensure that every tablet contains the amount of drug substance intended with little variation among tablets within a batch. 10 tablets were weighed and triturated. The tablet triturate equivalent to 50mg of the drug was weighed accurately, dissolved in pH 1.2 buffers and diluted to 100ml with the same. Further dilutions were done suitably to get a concentration of 10μg/ ml with simulated gastric fluid pH 1.2. Absorbance was read at 259nm against the reagent blank, and the concentrations of drug in μg/ ml was determined by using the regression equation.
Drug content in mg / tablet = conc. μg/ml * dilution factor /1000
% Drug content = drug content in mg * 100 / label claim.
In -vitro dissolution studies

In-vitro release studies were carried out using a modified USP XXIII dissolution test apparatus Lab India, DS-800). The dissolution fluid was 500ml of 6.8pH phosphate buffer at a speed of 50rpm at a temperature of 370c were used in each test. Samples of dissolution medium (5ml) were withdrawn for every 2min and assayed for Probenecid by measuring absorbance at 259nm. For all the tests 5ml of the test medium were collected at specified time intervals and replaced with same volume of 6.8pH phosphate buffer Figure 4.
Details
Apparatus used : USP II Lab India DS800 Dissolution Medium 6.8 pH phosphate buffer Dissolution Medium volume, 500ml Temperature 37oC
Speed of paddle : 50rpm
Sampling Intervals : 2, 4, 6, 8, 10, 15, 20, 25 and 30min Sample withdrawn 5ml
Absorbance measured : 259nm.
Results and discussion
Standard Calibration curve of Probenecid
It was found that the estimation of Probenecid by UV spectrophotometric method at λmax 259.0nm in 6.8pH phosphate buffer had good reproducibility and this method was used in the study. The correlation coefficient for the standard curve was found to be closer to 1 (Tables 3 & 4).


Evaluation Parameters for Fast Dissolving Tablets of Probenecid
Pre-compression parameters: The data were shown in Table 5 the values for angle of repose were found in the range of 25°-30°. Bulk densities and tapped densities of various formulations were found to be in the range of 0.41 to 0.50 (gm/ cc) and 0.50 to 0.58 (gm/cc) respectively. Carr's index of the prepared blends was fall in the range of 13.06% to 18.18%. The Hausner ration was fall in range of 1.14 to 1.22. From the result it was concluded that the powder blends had good flow properties and these can be used for tablet manufacture Figures 5 & 6.




Post compression Parameters:
a. Weight variation test: Tablets of each batch were subjected to weight variation test, difference in weight and percent deviation was calculated for each tablet and was shown in the Table 6. The average weight of the tablet is approximately in range of 407 to 398.5, so the permissible limit is ±5% (>250mg). The results of the test showed that, the tablet weights were within the pharmacopoeia limit.
b. Hardness test: Hardness of the three tablets of each batch was checked by using Monsanto hardness tester and the data's were shown in Table 6. The results showed that the hardness of the tablets is in range of 2.0 to 2.5kg/cm2, which was within IP limits [13,14].
c. Thickness: Thickness of three tablets of each batch was checked by using Vernier Caliper and data shown in Table 6 The result showed that thickness of the tablet is raging from 4.56 to 5.34.
d. Friability: Tablets of each batch were evaluated for percentage friability and the data were shown in the Table 6 The average friability of all the formulations lies in the range of 0.30 to 0.51% which was less than 1% as per official requirement of IP indicating a good mechanical resistance of tablets.
e. In vitro disintegration time: Tablets of each batch were evaluated for in vitro disintegration time and the data's were shown in the Table 6 The results showed that the disintegration time of prepared tablets were in the range of 17 to 25.33seconds.
Assay: Assay studies were performed for the prepared formulations. From the assay studies it was concluded that all the formulations were showing the % drug content values within 97.23-100.26%.
In-vitro Dissolution studies: In-vitro dissolution studies were carried out by using 500ml of 6.8pH phosphate buffer in USP dissolution apparatus by using paddle method. The dissolution studies were carried out for about 30min. The dissolution data for all the formulations were given in the Tables 5 & 6.
Conclusion
In the present work, an attempt has been made to develop rapidly disintegrating tablets of Probenecid. Novel method of co processed super disintegrates technology was employed to formulate the tablets. All the formulations were prepared by direct compression method. The blend of all the formulations showed good flow properties such as angle of repose, bulk density, tapped density. The prepared tablets were shown good post compression parameters and they passed all the quality control evaluation parameters as per I.P limits. Among all the formulations F4 formulation showed maximum drug release i.e., 98.16% in 30min hence it is considered as optimized formulation. The F4 formulation contains CP2 as super disintegrate in the concentration of 50mg. (CP 2 contains CCS and CP in 1:2 ratio).
References
- Velmurugan S, Sundar V (2010) Oral Disintegrating Tablets: An Overview. International Journal of Chemical and Pharmaceutical Sciences 1(2): 1-12.
- Yash Paul, Sarvan Tyagiand, Bhupinder Singh (2011) Formulation and Evaluation of Oral Dispersible Tablets of Zidovudine with different Super disintegrants. International Journal of Current Pharmaceutical Review and Research 2(2).
- Goodman and Gilman Manual of Pharmacology and Therapeutics, 2nd edn. By Randa Hilal Dandan, Laurence Brunton.
- Keny RV Desouza C (2010) Lourenco CF Formulation and evaluation of rizatriptan benzoate mouth disintegrating tablets. Indian J Pharm Sci 72(1): 79-85.
- Parikh BN, Patel DM, Patel CN, Dave JB, Gothi GD, et al. (2010) Formulation optimization and evaluation of immediate release tablet of telmisartan. J Global Pharm Tech 2(2): 79-84.
- Evren alğin yapar (2014) Orally Disintegrating Tablets: An Overview. J App Pharm Sci 4(2): 118-125.
- Shirsand SB, Raman G (2010) Novel co-processed super disintegrants in the design of fast dissolving tablets. International Journal of Pharma and Bio Sciences 1(1): 222-227.
- Vishwanadham Y, haritha G (2016) Development and validation of RP-HPLC method for simultaneous estimation of naproxen and esomeprazole in pharmaceutical dosage. JIPBS 3(3): 39-42.
- Divya A, Vishwanadham Y, Mounika (2017) Development and Validation of RP-HPLC Method for Simultaneous Determination of Diclofenac Sodium and Eperisone Hydrochloride in Pharmaceutical Dosage. Pharmaceutics analytical Acta 8(6): 1-6.
- Sreekanth D, Ramya P, Vishwanadham Y, Vanitha R (2017) Development and Method Validation of RP-HPLC For Simultaneous Determination of Pregabalin and Methylcobalamin in Pure and Pharmaceutical Dosage. Asian Journal of Research in Chemistry 10(4): 557-565.
- Vishwanadham Y, Divakar K, Hari A, Prakash Om, Kishore M, et al. (2016) world journal of pharmacological research and technology 4(5): 11-18.
- Vishwanadham Y, Kumaraswamy T, Appaji D (2015) World Journal of Pharmacy and Pharmaceutical Sciences 4(8): 1444-1451.
- Rapaka G, Haritha D, Sreekanth V (2017) Formulation development and invitro evaluation of matrix type transdermal drug delivery system using cetyl pyridinium. Int J Pharm 7(3): 51-56.
- Sandhya M, Vishwanadham Y (2016) formulation and evaluation of atorvastatin calcium sustained release tablets. Int J Pharm 6(3): 124 -130.















