




CRISPR/ Cas9: A Toolbox with Clinical Potential
Surya Bansi singh and Deepa Subramanyam*
National Centre for Cell Science, Savitribai Phule Pune University Campus, India
Submission: July 11, 2016; Published: August 05, 2016
*Corresponding author: Deepa Subramanyam, National Centre for Cell Science, Savitribai Phule Pune University Campus, Ganeshkhind, Pune - 411007, Maharashtra, India.
How to cite this article: Surya Bansi S, Deepa S. CRISPR/ Cas9: A Toolbox with Clinical Potential. Int J cell Sci & mol biol. 2016; 1(1): 555554. DOI:10.19080/IJCSMB.2016.01.555554
Abstract
The CRISPR/ Cas9 machinery, which is known to provide adaptive immunity to bacteria and archeae, has been proven an efficient tool for genome editing in a wide range of organisms. It is an RNA-guided DNA endonuclease that introduces double-stranded breaks in the target DNA. This compact machinery has been adapted and is being widely used for precise editing and modification of the genome [1]. This review focuses on genome editing using the CRISPR/ Cas9 system for correction of genetic diseases. Here, we review examples from literature where the CRISPR/ Cas9 system has been used for the correction of Duchenne Muscular Dystrophy (DMD), cataract caused due to mutations in the Crygc gene, cystic fibrosis, and sickle cell disease.
Keywords: Nuclease; Genome editing; Genetic disease; Duchenne muscular dystrophy (DMD); Cataract; Cystic fibrosis; Sickle cell disease
Abbreviations: CRISPRs: Clustered Regularly Interspaced Short Palindromic Repeats; tracrRNA: Trans-Activating RNA; PAM: Protospacer Adjacent Motif; DSBs: Double-Strand Breaks; NHEJ: Non-Homologous End Joining; HDR: Homology-Dependent Repair; sgRNA: Single-Guide RNA; AON: Antisense Oligonucleotide; hiPSC: Human Induced Pluripotent Stem Cell; CFTR: Cystic Fibrosis Transmembrane Regulator Protein
Introduction
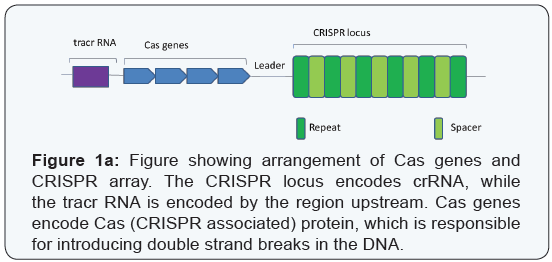
CRISPRs (clustered regularly interspaced short palindromic repeats) and the associated nuclease, Cas9, were originally discovered as components of an RNA-mediated adaptive immune system in prokaryotes [2-5]. The system comprises of a CRISPR RNA (crRNA) that shares sequence complementarity to the invading genetic material, and a trans-activating RNA (tracrRNA) that binds and targets the Cas9 endonuclease to a specific region of the DNA to mediate cleavage [5]. The crRNA is encoded by the CRISPR array, while the tracrRNA is encoded upstream of CRISPR-Cas locus (Figure 1a) [1,6]. This system has been tweaked to target specific sites in the genome, permitting editing of the same [1].
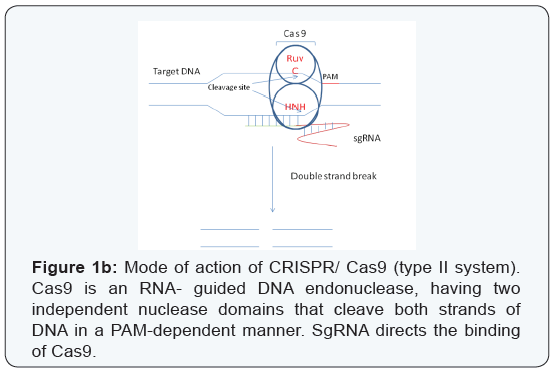
According to their structural and functional differences, the CRISPR/ Cas9 system has been classified into three major types - types I, II and III. The type I and type III systems use a large multi-protein complex for RNA-guided DNA cleavage [7], while the type II system uses a single Cas9 protein for DNA recognition and cleavage [1]. Both a seed sequence in the crRNA, and a conserved protospacer adjacent motif (PAM) sequence in the target are crucial for Cas9-mediated cleavage [1,8]. The type II system has been found to be the most adaptable and is currently widely used. Cas9 is an RNA-guided double-stranded DNAse containing two independent nuclease domains, namely RuvC and HNH, which together can cleave both strands of the target DNA resulting in double-strand breaks (DSBs) [1,8]. The crRNA together with tracr RNA form a complex that drive conformational changes in Cas9, which direct binding and cleavage in a PAM-dependent manner (Figure 1b) [9-11] .
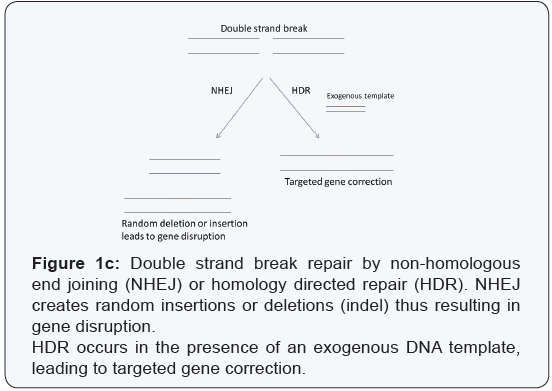
On being cleaved, the target DNA fragment can be repaired via either non-homologous end joining (NHEJ) or homologydependent repair (HDR) (Figure 1c). NHEJ is an error-prone pathway that can result in the introduction of indels (insertions or deletions) eventually leading to a frame shift mutation and a gene knockout. HDR depends on the presence of an exogenous template on the basis of which the DSB can be repaired. Both these mechanisms can be exploited to modify the genetic content in the region close to the DSBs generated by Cas9 [12-15]. This system of genetic editing has been further simplified and has been used in the rapid generation of genetically modified mice by injecting a single-guide RNA (sgRNA), which is a fusion of the tracrRNA and the crRNA along with mRNA encoding Cas9, into mouse embryos [16]. Due to the specificity, versatility, efficiency, simplicity, and multiplexing capability in engineering biological systems, the CRISPR/ Cas9 system can also be utilized in the correction of genetic diseases.
Genetic Diseases
Many genetic variations such as insertions, translocations, duplications, or deletions are commonly associated with human genetic disease. Single point mutations in the coding region of a gene are often associated with severe genetic diseases [17]. In the following sections, we review the recent literature focused on using the CRISPR/ Cas9 system for the correction of genetic diseases.
Muscular dystrophy
One of the most commonly occurring genetic diseases is Duchenne Muscular Dystrophy (DMD). DMD occurs due to a mutation in the dystrophin gene [18]. The mutation results in a loss of function of this important skeletal muscle protein which helps in maintaining the integrity of the muscle cell membrane during contraction, by linking the dystroglycan complex at the cell surface with the underlying cytoskeleton [19].
One of the earliest approaches for correction of this disease involved removal of the internal, but unessential region of the mutated dystrophin gene, thus restoring the proper reading frame of dystrophin gene [20]. In another approach, an antisense oligonucleotide (AON)- mediated exon skipping strategy was used, which masked splice donor or accepter sequences surrounding the mutated exon in the DMD mRNA, thereby restoring biological function of the dystrophin protein in mice [21,22] and humans [23-25]. A number of limitations are associated with this strategy, which include variable efficiency of uptake by tissues, repeated AON injections in order to maintain effective skipping, and toxicity associated with AON [26].
Till date, various designer nucleases (ZFNs and TALENs) have been used as a tool for correction of DMD by restoring the native dystrophin gene [27,28]. However, genome editing with ZFNs and TALENs have been shown to have occasional cytotoxic [29] or off-target effects [30,31].
With the advent of the CRISPR/ Cas9 system in the field of genome editing, accelerated progress in this area has been observed. The dystrophic phenotype in a mouse model of DMD was corrected through gene editing by injecting mRNA encoding Cas9, the specific sgRNA, and a single-stranded DNA oligonucleotide for correction (this served as a template for HDR) into single cell mouse embryos [32]. It has also been reported that an in-frame deletion of a region between exons 45–55, produces dysfunctional dystrophin, and is associated with the milder Becker muscular dystrophy [33,34].
Mutational hotspots spanning the region between exons 45–55 were targeted using sgRNAs and Cas9, to generate a deletion of 336 kb in skeletal myoblasts from DMD patients. This approach showed greater than 60% correction of existing mutations [35]. In another approach, the region between exons 45–55 were targeted using CRISPR/ Cas9-mediated NHEJ, to reframe dystrophin in human induced pluripotent stem cell (hiPSC) lines. Restoration of dystrophin and b-dystroglycan after engraftment of the reframed hiPSC- derived skeletal muscle cells into a mouse model of DMD was observed [36].
The occurrence of a premature termination codon in exon 23, which is responsible for the dystrophic phenotype of mdx mice, has been successfully targeted using CRISPR/ Cas9 to create an internal genomic deletion [37]. Targeted gene modifications and restoration of dystrophin expression have been shown in terminally differentiated skeletal muscle fibers and cardiomyocytes, as well as in muscle satellite cell (precursor of skeletal muscle cell), in both neonatal and adult mice. These mice show restored dystrophin expression and partial recovery from the deficiency [38]. Thus, CRISPR/ Cas9-mediated gene editing offers a ray of hope for patients suffering from DMD.
Cryg-dependent cataract
Cataract, which is characterized by opacity of the lens, is a common cause of blindness. Till date, nearly 140 mutations have been identified which are responsible for causing cataract in mice. The most frequently found mutation is in the Cryg gene family [39]. The Cryg gene, consisting of three exons, is conserved in all mammals, and belongs to the β- and γ- crystalline-encoding gene superfamily. All three exons collectively encode for a 21 kDa protein [40,41]. Mutations in the γ- crystalline gene are known to affect the normal development of the eye lens by promoting hyperproliferation of lens epithelia and forming polygonal epithelial cells. This creates a disturbance in the arrangement and shape of fiber cells, leading to the formation of cataract.
It was reported that a 1- bp deletion in the Crygc gene leads to a stop codon at the 76th amino acid in exon 3, which is known for encoding a beta sheet of the protein [39]. It was observed that there are a high number of mutations in cryg/ CRYG, in both mouse and humans respectively, which make this gene cluster a hot spot for autosomal dominant cataract [42-45].
Recently, an attempt has been made to correct this mutation via HDR using exogenously supplied oligos, or by NHEJ, mediated by the double strand break introduced by CRISPR/ Cas9. Different regions of the mutant cryg gene were targeted by sgRNAs, two of which were targeted to the 1-bp deletion in the mutant allele. Correction of cataract was seen at the organismal level by co-injecting the Cas9 mRNA and sgRNA into the cytoplasm of the zygote in mouse [46]. This study also reported successful germline transmission of the corrected allele to the next generation. Further studies using the CRISPR/Cas9 system are required for successful translation of this approach to correct cataract.
Cystic fibrosis
Cystic fibrosis is one of the most common chronic and life threatening genetic diseases. This disease affects the lungs, and is caused by a mutation in the CFTR gene leading to the failure of an epithelial cell chloride channel to respond to cAMP [47,48]. It is characterized by the production and accumulation of thick, sticky mucus in the lungs that impairs breathing and provides an environment for pathogens to flourish, resulting in respiratory failure. 70% of cystic fibrosis cases are characterized by production of a variant form of the protein, which includes deletion of phenylalanine 508 (ΔF 508) in exon 11. This causes protein misfolding, ER retention, and degradation of the cystic fibrosis transmembrane regulator protein (CFTR) [49-51].
In a recent study, organoids from the small and large intestine of two different pediatric cystic fibrosis patients have been established as model to study cystic fibrosis. These organoids were targeted with two different sgRNAs, together with a donor plasmid carrying the wild type CFTR sequence. Live cell microscopy showed the rapid expansion of corrected organoids which was not observed in the untransfected control organoids [52].
Further, the ability to generate pluripotent stem cells from accessible tissue from patients provides an excellent model system for human disease. iPSCs generated from a cystic fibrosis patient were corrected using CRISPR/ Cas9, followed by differentiation into mature airway epithelial cells. These cells demonstrated normal expression and function of CFTR, furthering the belief that gene editing using CRISPR/ Cas9 may help correct similar gene defects in patients [53].
Sickle cell disease
Sickle cell disease is an inherited monogenic disorder caused by a mutant allele of β-globin, called sickle hemoglobin (HbS). A point mutation in the β- globin gene, results in malformation of the hemoglobin structure, leading to abnormal sickle-shaped RBCs. This disease affects approximately 300,000 neonates every year [54]. Inheriting two abnormal copies of the β- globin gene, is the underlying genetic basis of this disease [55].
The earliest approach to treat this disease involved the delivery of γ- globin or the antisickling β- globin protein to inhibit polymerization of the variant HbS [56,57] More recently CRISPR/ Cas9 has been used for the correction of this disease in human iPSCs, derived from adult sickle cell disease patients. These corrected stem cells were later differentiated into erythrocytes and were shown to produce β- globin protein from the corrected allele [58]. These and other studies represent a step towards using gene editing to potentially cure genetic diseases.
CRISPR/Cas9: Promise for the Future
Genome editing holds huge promise in the field of genetic disorders and cancer. The relative simplicity of the CRISPR/Cas9 system makes it extremely attractive. Perhaps not too long from now in the future, one may hope that research will advance to the stage where we will be able to correct relatively simpler genetic disorders that arise due to single point mutations in a specific gene. More importantly, the CRISPR/ Cas9 system can be used to model genetic diseases by engineering specific mutations in either cell lines or model organisms. These can then be used for screening drugs or treatment strategies that can potentially be taken back to the clinic. These tools can even be used in the context of more complicated genetic disorders such as cancer. However, caution must still be exercised, as the CRISPR/ Cas9 system can potentially have off-target effects.
Acknowledgement
DS is a recipient of an Intermediate Fellowship from the Wellcome Trust -Department of Biotechnology (DBT) India Alliance.
References
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, et al. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096): 816-821.
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, et al. (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315(5819): 1709-1712.
- Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169(12): 5429-5433.
- Makarova KS, Grishin NV, Shabaline SA, Wolf YI, Koonin EV (2006) A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biology Direct 1: 7.
- Marraffini LA, Sontheimer EJ (2010) CRISPR interference: RNAdirected adaptive immunity in bacteria and archaea. Nature Reviews Genetics 11(3): 181-190.
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, et al. (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471(7340): 602-607.
- Makarova KS, Aravind L, Wolf YI, Koonin EV (2011) Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol Direct 6: 38.
- Gasiunas G, Barrangou R, Horvath P, Siksnys V (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA 109(39): E2579-E2586.
- Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, et. al. (2014). Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343(6176): 1247997.
- Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, et. al. (2014) Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156(5): 935-949.
- Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507(7940): 62-67.
- Cong L, Ran FA, Cox D, Lin S, Barretto R, et al. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121): 819-823.
- Cho SW, Kim S, Kim JM, Kim JS (2013) Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31: 230-232.
- Jinek M, East A, Cheng A, Lin S, Ma E, et al. (2013) RNA-programmed genome editing in human cells. Elife 2: e00471.
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, et al. (2013) RNA-guided human genome engineering via Cas9. Science 339(6121): 823-826.
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, et al. (2013) One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell 153(4): 910-918.
- Stankiewicz P, Lupski JR (2010) Structural variation in the human genome and its role in disease. Annu Rev Med 61: 437-455.
- Hoffman EP, Brown RH, Kunkel LM (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51(6): 919- 928.
- Campbell KP, Kahl SD (1989) Association of dystrophin and an integral membrane glycoprotein. Nature 338(6212): 259-262.
- Lu QL, Yokota T, Takeda S, Garcia L, Muntoni F, et al. (2011) The status of exon skipping as a therapeutic approach to duchenne muscular dystrophy. Mol Ther 19(1): 9-15.
- Lu QL, Mann CJ, Lou F, Bou-Gharios G, Morris GE, et al. (2003) Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat Med 9(8): 1009-1014.
- Echigoya Y, Aoki Y, Miskew B, Panesar D, Touznik A (2015) Long-term efficacy of systemic multiexon skipping targeting dystrophin exons 45-55 with a cocktail of vivo-morpholinos in mdx52 mice. Mol Ther Nucleic Acids 4(2): e225.
- Van Deutekom JC, Janson AA, Ginjaa IB, Frankhuizen WS, Aartsmarus A, et al. (2007) Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med 357(26): 2677-2686.
- Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, et al. (2009) Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol 8(10): 918-928.
- Aartsma-Rus A, Fokkema I, Verschuuren J, Ginjaar I, van Deutekom J, et al. (2009) Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat 30(3): 293- 299.
- Goyenvalle A, Griffith G, Babbs A, Andaloussi SEI, Ezzat K, et al. (2015) Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat Med 21(3): 270-275.
- Ousterout DG, Perez-Pinera P, Thakore PI, Kabadi AM, Brown MT, et al. (2013) Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol Ther 21(9): 1718-1726.
- Rousseau J, Chapdelaine P, Boisvert S, Almeida LP, Corbeil J, et al. (2011) Endonucleases: tools to correct the dystrophin gene. J Gene Med 13(10): 522-537.
- Cornu TI, Thibodeau-Beganny S, Guhl E, Alwin S, Eichtinger M, et al. (2008) DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol Ther 16(2): 352-358.
- Guilinger JP, Pattanayak V, Reyon D, Tsai SQ, Sander JD, et al. (2014) Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat Methods 11(4): 429-435.
- Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, et al. (2008) Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nature Biotechnology 26(7): 808- 816.
- Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, et al. (2014) Prevention of muscular dystrophy in mice by CRISPR/ Cas9- mediated editing of germline DNA. Science 345(6201): 1184-1188.
- Béroud C, Tuffery-Giraud S, Matsuo M, Hamroun D, Humbertclaude V, et al. (2007) Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum Mutat 28(2): 196-202.
- Taglia A, Petillo R, D’Ambrosio P, Picillo E, Torella A, et al. (2015) Clinical features of patients with dystrophinopathy sharing the 45-55 exon deletion of DMD gene. Acta Myol 34(1): 9-13.
- Ousterout DG, Kabadi AM, Thakore PI, Majoros WH, Reddy TE, et al. (2015) Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun 6: 6244.
- Young CS, Hicks MR, Ermolova NV, Nakano H, Jan M, et al. (2016) A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell 18(4): 533-540.
- Long C, Amoasii L, Mireault AA, McAnally JR, Li H, et al. (2016) Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351(6271): 400-403.
- Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, et al. (2016) In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351(6271): 407-411.
- Zhao L, Bao S, Zhao G, Liang Y, Li K, et al. (2010) Mutation, distribution, positional cloning for cataract in house mouse. Chin J Comput Med 20(7): 62-66.
- Graw J (1997) The crystallins: genes, proteins and diseases. Biol Chem 378(11): 1331-1348.
- Slingsby C, Clout NJ (1999) Structure of the crystallins. Eye Lond 13 ( Pt 3b): 395-402.
- Graw J, Klopp N, Lo¨ster J, Soewarto D, Fuchs H, et al. (2001) ENUinduced mutation in mice leads to the expression of a novel protein in the eye and to dominant cataracts. Genetics 157(3): 1313-1320.
- Graw J, Neuba¨user-Klaus A, Klopp N, Selby PB, Lo¨ster J, et al. (2004) Genetic and allelic heterogeneity of Cryg mutations in eight distinct forms of dominant cataract in the mouse. Invest Ophthalmol Vis Sci 45(4): 1202-1213.
- Klopp N, Favor J, Lo¨ster J, Lutz RB, Neuha¨user-Klaus A, et al. (1998) Three murine cataract mutants (Cat2) are defective in different c-crystallin genes. Genomics 52(2): 152-158.
- Li L, Chang B, Cheng C, Chang D, Hawes NL, et al. (2008) Dense nuclear cataract caused by the gamma B-crystallin S11R point mutation. Invest Ophthalmol Vis Sci 49(1): 304-309.
- Wu Y, Liang D, Wang Y, Bai M, Tang W, et al. (2013) Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 13(6): 659-62.
- Frizzell RA, Rechkemmer G, Shoemaker RL, (1986) Altered regulation of airway epithelial cell chloride channels in cystic fibrosis. Science 233(4763): 558-560.
- Li M, McCann JD, Liedtke CM, Nairn AC, Greengard P, et al. (1988) Cyclic AMP-dependeny protein kinase opens chloride channels in normal but not cystic fibrosis airway epithelium. Nature 331(6154): 358-360.
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, et al. (1989) Identification of the cystic fibrosis gene: genetic analysis. Science 245(4922): 1073-1080.
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, et al. (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245(4922): 1066-1073.
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, et al. (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63(4): 827-834.
- Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, et al. (2013) Functional Repair of CFTR by CRISPR/Cas9 in Intestinal Stem Cell Organoids of Cystic Fibrosis Patients. Cell Stem Cell 13(6): 653-658.s
- Firth AL, Menon T, Parker GS, Qualls SJ, Lewis BM, et al. (2015) Functional Gene Correction for Cystic Fibrosis in Lung Epithelial Cells Generated from Patient iPSCs. Cell Reports 12(9): 1385-1390.
- Piel FB (2016) The present and future global burden of the inherited disorders of hemoglobin. Hematol Oncol Clin North Am 30(2): 327- 341.
- Ashley-Koch A, Yang Q, Olney RS (2000) Sickle hemoglobin (Hb S) allele and sickle cell disease: a HuGE review. Am J Epidemiol 151(9): 839-845.
- Pawliuk R, Westerman KA, Fabry ME, Payen E, Tighe R, et al. (2001) Correction of sickle cell disease in transgenic mouse models by gene therapy. Science 294(5550): 2368-2371.
- Levasseur DN, Ryan TM, Pawlik KM, Townes TM (2003) Correction of a mouse model of sickle cell disease: lentiviral/antisickling β-globin gene transduction of unmobilized, purified hematopoietic stem cells. Blood 102(13): 4312-4319.
- Huang X, Wang Y, Yan W, Smith C, Ye Z, et al. (2015) Production of gene-corrected adult beta globin protein in human erythrocytes differentiated from patient iPSCs after genome editing of the sickle point mutation. Stem Cells 33(5): 1470-1479.