




Comparative In-Vitro Dissolution Studies for Determination of Cefixime in an Innovator Product of Suprax Powder for Oral Suspension Dosage form Using Rp-Hplc Method
Mohamed EM Hassouna1* and Mahmoud A Mohamed2
1Chemistry Department, Faculty of Science, Beni-Suef University, Egypt
2HIKMA group, Pharmaceutical Company, Beni-Suef, Egypt
Submission: March 22, 2018; ; Published: April 04, 2018
*Corresponding author: Mohamed EM Hassouna, Chemistry Department, Faculty of Science, Beni-Suef University, Egypt, Tel: +201223861504; Email: Mohamed.hassona@science.bsu.edu.eg
How to cite this article: Mohamed E M Hassouna, Mahmoud A Mohamed. Comparative In-Vitro Dissolution Studies for Determination of Cefixime in an Innovator Product of Suprax Powder for Oral Suspension Dosage form Using Rp-Hplc Method. Glob J Oto, 2018; 14(3): 555886. DOI: 10.19080/GJO.2018.13.555886.
Abstract
Dissolution testing is an in vitro technique of great importance in formulation and development of all solid oral dosage forms and is used in all phases of development for product release and stability testing. The aim of this work is to evaluate and compare the different official and unofficial dissolution tests and treatments of obtained data by using J2 to determine whether all the formulations used are equivalent or significantly different. Accurate, simple and specific RP-HPLC method is performed for the determination of cefixime (CFX) in powder for oral suspension dosage form (POS) using FDA buffer media 0.05 M phosphate buffer, pH 7.2 is conducted for 12 doses from each brand for 45min. by using dissolution testing apparatus USP type-II (Paddle). Samples are withdrawn at time intervals of 10, 20, 30 and 45 minutes, then are analyzed for drug content by using HPLC technique.
Cumulative drug release at each time interval is calculated for POS and the obtained data are calculated with statistical techniques to meet the FDA requirements for obtaining a waiver of bioavailability and bioequivalence studies. The method is evaluated on Supleco LC-18-DB (4.6 mm x 250 mm) 5μm or equivalent, using mobile phase as a mixture of 0.04 mM tetra butyl ammonium hydroxide: acetonitrile (72.5:27.5 v/v) adjusted to pH 7.0 with 10% ortho phosphoric acid at a flow rate of 2.0 mL/min with UV detection at 254 nm. The injection volume is 10 μL, the retention time of cefixime is 3.195±0.679 min and the total run time is 6 min. The method has been applied for Aerocef 100 mg/5 mL POS and hence it is considered to be equivalent to the innovator product Suprax 100 mg/5mL using FDA media and the two products are considered similar.
Keywords: Cefixime Trihydrate; In Vitro Dissolution; Rp- Hplc; Stability Indicating Method; Generic Product; Innovator Product.
Abbreviations: CFX: Cefixime Trihydrate ; IVIVC: In vitro-In vivo Correlations; PTFPE: Polytetra- Fluorethylene; POS: Powder for Oral Suspension; FDA: US Food and Drug Administration; f1: The Difference factor; f2: Similarity factor.
Introduction
Dissolution testing is an in vitro technique can be used as a substitute for in vivo studies under strictly defined and specified conditions [1]. For the comparison of in vitro dissolution data and for use in vivo bioequivalence testing and in vitro-in vivo correlations (IVIVC), dissolution testing should be carried out under physiochemical and hydro dynamically defined conditions so that the obtained results are accurate and reproducible [1]. CFX, (Figure 1) is a semi synthetic cephalosporin antibiotic for oral administration. It is chemically designated as (6R, 7R)-7- [[(Z)-2-(2- Aminothiazol-4-yl)-2-[(carboxymethoxy) imino] acetyl] amino]-3- ethenyl-8-oxo-5-thia-1-azabicyclo [4.2.0] oct- 2-ene-2-carboxylic acid trihydrate. It has a molecular formula of C16H15N5 07 S2. 3H20 and a molecular weight of 507.5 [2]. CFX as an antibiotic is a third-generation cephalosporin like ceftriaxone and cefotaxime. CFX is highly stable in the presence of beta-lactamase enzymes. Thus, many organisms resistant to penicillin's and some cephalosporins due to the presence of beta-lactamases may be susceptible to CFX. Its antibacterial effect results from inhibition of mucopeptide synthesis in the bacterial cell wall. CFX is an orally absorbed third generation cephalosporin with a broad spectrum of activity against Gram- positive and Gram-negative bacteria and is highly resistant to beta-lactamase degradation. In general serum and urinary concentrations of CFX achieved after recommended daily doses are well above the minimal inhibitory concentrations at 90% for indicated microorganisms.

The dissolution test for immediate or controlled release in solid oral dosage forms has recently widened to a variety of novel or special dosage forms such as suspensions, chewing gums, transdermal patches, implants and others. Because of the different characteristics of early dosage form, the site absorption and dosing routes and applications, it is essential to give appropriate consideration to the following factors in the development of the test method: apparatus selection, dissolution medium, agitation and temperature [3]. CFX is official in British Pharmacopeia (BP) [4]. European Pharmacopeia (EP) and United States Pharmacopeia (USP) [5]. They include the HPLC method for its estimation. It is still a limited number of analytical methods that were reported for the determination of CFX individually or in combination with other drugs including colorimetric and spectrophotometric [6-15], capillary electrophoresis and electrochemical methods [16-19], thin layer chromatography (TLC) [20,21], high performance liquid chromatography (HPLC) [22-33] and in vitro dissolution studies have been developed for the determination of CFX in capsule dosage forms using UV-VIS Spectrophotometer [34]. According to the best of our knowledge there is no in vitro dissolution study using HPLC method for determination CFX in Suprax powder for oral suspension dosage form by using FDA dissolution media. Consequently, the aim of this work was to establish and compare the obtained data by using J2 for official and unofficial products.
Materials and Reagents
Raw Materials and Reagents
Cefixime is kindly supplied as C16H15N5O7S2.3H2O, Molecular Weight: 507.50 by Hikma pharmaceutical industries company, Beni-Suef, Egypt with claimed purity of 100.43%. Innovator Product Suprax® POS (Batch No. 2099) is manufactured by Hikma pharmaceutical industries company, Beni-Suef, Egypt. Each 5 mL are claimed to contain 100 mg of CFX. Generic product Aerocef POS (Batch No. 2000) is manufactured by Riva Parma Company, 6th October, Egypt. Each 5mL are claimed to contain 100 mg of CFX. Xanthan Gum is supplied by Semiramis international trading, Sodium benzoate was supplied by Tenc Zhou Tenc Long Chemical Co., LTD, Strawberry powder flavour was supplied by Givaudan and Sucrose crystalline is supplied by Tereos pharmaceutical excipient company. Acetonitrile, methanol HPLC-grade, 40% tetra butyl ammonium hydroxide solution, purified water, potassium dihydrogen orthophosphate, di-sodium hydrogen phosphate anhydrous, sodium hydroxide, hydrochloric acid, hydrogen peroxide and ortho phosphoric acid analytical grades, are procured from (Scharlau, Spain).
Equipment and chromatographic conditions
Chromatographic separation of CFX is performed at room temperature in an HPLC (Shimadzu LC SPD 20 A) with a detector (dual wavelength), equipped with binary pumps, auto sampler, oven CT0-20A/20AC with temperature range (10-85 °C) and LC Solution software. Chromatography is carried out under isocratic conditions with a mobile phase consisting of a mixture of 0.04 mM tetra butyl ammonium hydroxide: acetonitrile (72.5:27.5 v/v) adjusted to pH 7.0 with 10% ortho phosphoric acid at a flow rate of 2.0 mL/min and UV detection at 254 nm with injection volume of 10 μL using ACE C18 (4.6 mm x 250 mm) 5μm or equivalent and the total run time is 6 min. Dissolution apparatus (DIS 6000, Dr. Schleuniger, Germany) is used to study the drug dissolution profile. pH meter Mettler Toledo is used for the adjustment of the mobile phase and buffer solution.
Preparation of solvent, stock solutions and calibration standards
Solvent is prepared by transferring 6.8mL of concentrated phosphoric acid to 1000mL volumetric flask, addition of 300 mL of water, adjusting to pH 7.0±0.1 with 10 N NaOH then dilution to volume with water.
Stock standard solutions of cefixime (1115pg mL-1)
Accurately transfer 111.5 mg of the standard CFX (equivalent to 100 mg cefixime base) into 100-ml volumetric flask, add about 70 ml diluents, sonicate for 10 min till complete dissolution then complete to volume with the same diluents. Filter through suitable syringe filter.
Working standard solutions of Cefixime (111.5pg mL-1)

Accurately transfer 10 mL from the stock standard solution of CFX into 100 mL volumetric flask, add diluents and sonicate to dissolve. Makeup to the mark with the same diluents and mix well. Inject into the chromatographic system. The chromatogram obtained is shown in (Figure 2).
Calibration curves Construction
Different aliquots of CFX equivalent to 27.87-223μg/mL are separately transferred from their respective stock standard solutions into separate series of 100mL volumetric flasks and volume is completed to the mark with diluents and mixed well. Triplicate 10μL injections are performed for each concentration maintaining the flow rate at 2.0mL/min and the effluent is UV- scanned at 254nm. The chromatographic separation is carried out following the procedure under chromatographic conditions. The chromatograms are recorded and the peak areas of CFX are determined and the calibration curves relating the obtained integrated peak areas to their corresponding concentrations are constructed and the regression equations are computed.
Application to pharmaceutical formulation (Suprax®100 mg POS and Aerocef® 100 mg POS)
Constitute Suprax as directed in the labeling, dilute 5mL (equivalent to about 100mg of cefixime) in 500mL volumetric flask. Sonicate if necessary to ensure complete dissolution of cefixime. Transfer 10mL into 20mL volumetric flask to obtain a final concentration of (111.5μgmL-1). Filter through 0.45μm PTFE membrane filter to obtain the clear assay preparation.
Content Uniformity
The test for content uniformity is required for all dosage forms not meeting the conditions for the mass variation test. Alternatively, products that do not meet the 25mg/25 per cent threshold limit may be tested for uniformity of dosage units by mass variation instead of the content uniformity test on the following condition: the concentration Relative Standard Deviation (RSD) of the active substance in the final dosage units is not more than 2 per cent, based on process validation data and development data, and if there has been regulatory approval of such a change. The concentration RSD is the RSD of the concentration per dosage unit (m/m or m/V), where concentration per dosage unit equals the assay result per dosage unit divided by the individual dosage unit mass [2].
In Vitro Dissolution Studies
Dissolution Parameters:
a) Apparatus: USP II (paddle type).
b) Speed: 50rpm.
c) Medium: 900 mL FDA dissolution medium.
d) Temperature: 37.0±0.5 °C.
e) Time: 25mL sample aliquots are withdrawn at 10, 20, 30 and 45 min using a hypodermic glass syringe equipped with a stainless-steel needle, and replaced with equal volume of the fresh medium at the same temperature to maintain constant total volume. The solutions are immediately filtered through a 0.45 μm membrane. Drug release is assayed by HPLC method. Cumulative percentages of the dissolved drug are calculated and plotted versus time.
Dissolution Medium Preparation (phosphate buffer pH 7.2): Dissolve 81.6 g of anhydrous potassium dihydrogen phosphate in 12.0 liters with de ionized water so as to obtain 0.05M solution. If necessary, adjust the pH to 7.2 ± 0.05 with 1N phosphoric acid or 1N NaOH. For mobile phase and chromatographic conditions; proceed as directed under assay method.
Preparation of Stock Standard Solution for Dissolution Test (1115μg mL-1): Similar to the previously prepared solution, accurately transfer 111.5 mg of the standard CFX (equivalent to 100 mg cefixime base) into 100-mL volumetric flask, with the aid of 20 mL methanol, sonicate for about 5 minutes and complete to volume with the pH 7.2 buffer solution and mix well. Filter through suitable syringe filter.
Working standard solutions of cefixime (111.5 mL-1): Accurately transfer 10mL from the stock standard solution of CFX into 100mL volumetric flask, add the dissolution medium and sonicate to dissolve. Makeup to the mark with the same diluents and mix well. Inject into the chromatographic system. The obtained chromatogram is shown in (Figure 3).

Test Preparation for Dissolution of (Suprax®100 mg POS and Aerocef ® 100 mg POS):
Place 12 doses each in a vessel from Aerocef ® 100 mg POS the generic product and Suprax®100 mg POS, the innovator product. Operate using the above conditions. After 10, 20, 30 and 45 minutes period.
Results and Discussion
Methods development and optimization
Mobile phase and wavelength selection: Many systems of different compositions and ratios were tried including: methanol (100%); methanol: purified water (70:30, v/v); acetonitrile: purified water (70:30, v/v) and methanol: purified water (95:5, v/v). All the solvents of the mobile phase were filtered through 0.45μ membrane to remove particulate matter and degassed by sonication; also (0.8, 1.0, 1.5 and 2.0mL/min) flow rates were tried, to get the optimum wavelength for the CFX solution, a solution of 40 μg/mL of CFX was measured at 200 to 400nm (Figure 4). Thus, 254nm was selected as the most suitable wavelength. Preliminary studies involved trying C18 reversed- phase columns. The best developing system was the mobile phase composed of a mixture of 0.04mM tetra butyl ammonium hydroxide: acetonitrile (72.5:27.5 v/v) adjusted to pH 7.0 with 10% ortho phosphoric acid at a flow rate of 2.0mL/min and UV detection at 254nm. Another advantage is the relatively short run time (6min) of the analysis, under isocratic conditions. This selected developing system allows good separation with good Rt values without tailing of the separated bands and good theoretical plates.

Optimization of dissolution test conditions
Dissolution is an official test used by pharmacopoeias for drug evaluation release of solid and semisolid dosage forms, and it is routinely used in Quality Control (QC) and Research & Development (R&D) laboratories. The purpose of in vitro dissolution studies in QC is batch to batch consistency and detection of manufacturing deviation while in R&D the focus is to provide some predictive estimate of the drug release in respect to the in vivo performance of a drug product. For QC, an over-discriminatory test might be suitable to detect even small production deviations. However, for prediction of the in vivo performance of drug product, a dissolution test should be sensitive and reliable [35]. The accomplishment of dissolution profile is recommended as a support in the development and optimization of drug formulation as well as in the establishment of in vitro/in vivo correlation. When dissolution test is not defined or if the monograph is not available, comparison of drug dissolution profiles is recommended on three different dissolution mediums (pH 1-7.5) [36]. In vitro dissolution is used to perform the release rate of drug products and to assure the quality of suspension dosage forms by the pharmaceutical industry and regulatory agencies [37].
Method validation
The method was validated, in accordance with ICH guidelines (ICH Q2R1), for system suitability, precision, accuracy, linearity, specificity, ruggedness, robustness, LOD and LOQ [38].
Linearity and range
The linearity of the proposed methods is obtained in the concentration range (55.75 - 446μg/mL) for CFX at FDA buffer media of pH 7.2. From the graphical presentation, the relation of peak response (A) plotted at X axis and concentration (C) plotted at Y axis is evaluated on the base of least-square linear regression equation (A= slope C + Y intercept). The LOD and LOQ of the method are calculated using the expression (3.3a/ slope) and (10a/slope) respectively. The obtained coefficient of regression is 0.9999. Linearity results are shown in (Table 1).

aLimit of detection (3.3* o /Slope) and limit of quantization (10* o / Slope).
Repeatability
Repeatability is conducted using 6 replicates of the standard solution of the compound being studied (111.5 ng/mL). The system is precise as the relative standard deviation RSD 2%. Precision of the method is illustrated in (Table 1).
Accuracy and recovery

The recovery was calculated for triplicate of three concentrations (55.75, 111.5 and 167.25μg/mL) comparing the individual peak response with that of the reference solution. Table 2 showed the recovery percent of 99.53±0.78 of the spiked analyte.
Formulation assay
The study is performed by assaying six samples from the finished product with the same concentration prepared from a homogenous sample starting from the first step of the analysis procedure to the final one. The results of the assay indicate that the method is selective for the analysis of Suprax® and Aerocef® Cap without interference from the excipients used to formulate. The results are displayed in (Table 3).

Intermediate precision (ruggedness)
Intermediate precision expresses the within-laboratories variations: different days, different analysts, different equipments, etc. Good results are obtained and presented in (Table 4).

Robustness
The method's capability to remain unaffected by small but deliberate variations in method parameters as: influence of variations of the pH in the mobile phase, influence of variations in its composition, different columns (different lots and/or suppliers), temperature, flow rate and wave length. Table 5 indicates the robustness of the proposed method.

System suitability
System suitability testing is an integral part of much analytical procedure. The tests are based on the concept that the equipments, electronics, analytical operations and samples to be analyzed constitute an integral system that can be evaluated as such. System suitability is checked by calculating the tailing factor (T), column efficiency (N), resolution (Rs) factors. All calculated parameters are found to be within the acceptable limits indicating good selectivity of the method and assuring system performance (Table 6).

Stability of the analytical solution
After preparing the standard solution, part of it was stored in fridge and another one was stored at room temperature. After about 24 hours, these solutions were tested against freshly prepared standard; relative standard deviation should be less than 2%. These results are displayed in (Table 7).

Potency of CFX standard=100.43%.
Content Uniformity
Select not less than 30 units, and proceed as follows for the dosage form designated. Where different procedures are used for assay of the preparation and for the content uniformity test, it may be necessary to establish a correction factor to be applied to the results of the latter. Assay 10 units individually using an appropriate analytical method. Calculate the acceptance value (AV) using the formula:
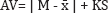
Where, T: label claim (100.0%), unless otherwise specified in the individual monograph,
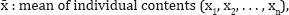
x1, x2, . . . , xn: individual contents of the units tested, expressed as a percentage of the
label claim,
n: sample size (number of units in a sample),
k: acceptability constant, k=2.4 when the sample size is 10, and k=2.0 when the sample
size is 30.
s: standard deviation of the sample = cf. (Table 8).


RSD: Relative Standard Deviation of the active substance in the final dosage units is not more than 2 per cent, based on process validation and development data, and if there has been regulatory approval of such a change = RSD results are illustrated in (Table 9).
Application of In Vitro Dissolution of Powder for oral Suspension dosage form
A simple model independent approach uses a difference factor (J1) and a similarity factor
(f2) is applied to compare the dissolution profiles. The difference factor (J1) calculates the
percent (%) difference between the two curves at each time point and is a measurement of the relative error between them. The equation of the similarity factor proposed by Moore and Flanner [39] is presented in equations (1, 2).
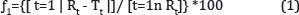
Where n is the number of time points, Rt is the dissolution value of the reference
(pre-change) batch at time t and Tt is the dissolution value of the test (post change) batch at time t. The similarity factor (f2) is a logarithmic reciprocal square root transformation of the sum of squared error and is a measurement of the similarity in the percent (%) dissolution between the two curves.
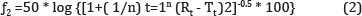
A specific procedure to determine the difference and similarity factors is as follows:
Determine the dissolution profile of the two products (12 units each) of the test (post change) and reference (pre-change) products using the mean dissolution values from both curves at each time interval. Calculate the difference (J1) and similarity Q2) factors using the above equations. Results are summarized in (Table 10). To obtain the in vitro dissolution profile of the product, the cumulative percentage of Aerocef and Suprax released is plotted against time (Figure 5).


Specificity
Method specificity was determined for the samples of the compound being studied and placebo matrix containing all excipients of the finished product. No interference was detected at the retention time of CFX in the placebo solution.
Conclusion
Successful in vitro dissolution studies have been applied for Aerocef 100mg/5mL powders for oral suspension and hence it is considered as equivalent to the innovator product Suprax 100mg/5mL powders for oral suspension at FDA dissolution media. The proposed RP-HPLC method for the determination of CFX in dosage forms is simple, precise, specific and accurate. The results of stress testing that have been undertaken according to the International Conference on Harmonization (ICH) guidelines revealed that the analytical method is valid, fit for use and can be applied for regular routine analysis and stability study.
Acknowledgement
The authors are thankful to the Publishers of the Global Journal of Otolaryngology for the provided publication gift. Also, they gratefully acknowledge Hikma pharmaceutical industries company, Beni-Suef, Egypt for providing cefixime and the Innovator Product Suprax® POS.
References
- Karande AD, Yeole PG (2006) Comparative Assessment of Different Dissolution Apparatus for Floating Drug Delivery Systems. Dissolution Technologies 20-23.
- British Pharmacopoeia (2017) Stationary Office, Medicines and Healthcare Products Regulatory Agency, London 2.
- Mamzoridi K, Kasteridou N, Peonides A, Niopas I (1996) Pharmacokinetics of Cefixime in Children with Urinary Tract Infections after a Single Oral Dose, Pharmacol Toxicol 78(6) : 417-420.
- Rivera Leyva JC, Garci'a Flores M, Valladares Mendez A, Orozco Castellanos LM, Martinez, et al. (2012) Comparative Studies on the Dissolution Profiles of Oral Ibuprofen Suspension and Commercial Tablets using Biopharmaceutical Classification System Criteria. Indian J Pharm Sci 74(4): 312-318.
- The United States Pharmacopoeia (2015) 38th Revision, NF 33, the United States Pharmacopoeia Convention Inc.
- Shah V, Raj H (2012) Development and Validation of Derivative Spectroscopic Method for Simultaneous Estimation of Cefixime Trihydrate and Azithromycin Dihydrate In Combined Dosage Form. International Journal of Pharma and Bio Sciences 3: 14-25.
- Sutar SV, Sancheti H, Patil SS (2013) Spectrophotometric method for cefixime trihydrate by using hydrotropic agent. International Science Press (India) 6(2): 80-85.
- Azmi SNH, Iqbal B, Al Mamari JK, Al Hattali KA, Al Hadhrami WN (2014) Method Development and validation for the determination of cefixime in pure and commercial dosage forms by spectrophotometer. Int J Chem Molec Nucl Mat Metallurg Eng 8(6): 595-601.
- Patel SA, Patel JV (2013) Spectrophotometric method for simultaneous estimation of cefixime trihydrate and linezolid in tablet dosage form. International Research Journal of Pharmacy 4(1): 161-164.
- Pekamwar SS, Kalyankar TM, Tambe BV, Wadher SJ (2015) Validated UV-Visible Spectrophotometric method for simultaneous estimation of cefixime and moxifloxacin in pharmaceutical dosage form. J Appl Pharm Sci 5(1): 37-41.
- Ramadan AA, Mandil A, Dahhan M (2013) UV-VIS Spectrophotometric study for determination of cefixime in pure form and in pharmaceuticals through complexation with Cu (II) using acetate-NaOH buffer in water: methanol. International Journal of Pharmacy and Pharmaceutical Sciences 5(1): 428-433.
- Anees MI, Baig MS, Tathe A (2015) Determination of cefixime and moxifloxacin in pharmaceutical dosage form by simultaneous equation and area under curve UV-spectrophotometric method. World Journal of Pharmacy and Pharmaceutical Sciences 4: 1172-1179.
- Shreya RS, Prasanna P, Suddhasattya D (2013) Quantitative estimation of cefixime and moxifloxacin in pharmaceutical preparation by UV spectrophotometric method. Int J Pharm Tech Res 5(1): 198-204.
- Kumar R, Singh P, Singh H (2011) Development of colorimetric method for the analysis of pharmaceutical formulation containing both ofloxacin and cefixime. International Journal of Pharmacy and Pharmaceutical Sciences 3(2): 178-179.
- Hassouna MEM, Abdelrahman MA, Mohamed MA (2017) Novel Spectro photometric Methods for Simultaneous Determination of Cefixime trihydrate and Sodium benzoate in Powder for Oral Suspension Dosage form. Glob J Oto 12(4): 555-841.
- Ahmed OA (2013) Simultaneous determination of ofloxacin and cefixime in tablet formulation using capillary electrophoresis. J Liq Chromatogr & Related Technol 36: 36-41.
- Afkhami A, Felehgari FS, Madrakian T (2013) Gold nano particles modified carbon paste electrode as an efficient electrochemical sensor for rapid and sensitive determination of cefixime in urine and pharmaceutical samples. Electrochimica Acta 103(3): 125-133.
- Babolia MA, Mani Varnosfaderani A (2014) Rapid and simultaneous determination of tetracycline and cefixime antibiotics by means of gold nanoparticles-screen printed gold electrode and chemo metrics tools. Measurement 47: 145-149.
- Jain R, Gupta VK, Jadon N, Radhapyari K (2010) Voltametric determination of cefixime in pharmaceuticals and biological fluids. Analytical Biochemistry 407(1): 79-88.
- Khandagle KS, Gandhi SV, Deshpande PB, Kale AN, Deshmukh PR (2010) High Performance Thin Layer Chromatographic determination of Cefixime and Ofloxacin in combined tablet dosage form. J Chem Pharm Res 2(5): 92-96.
- Rao J, Sethy K, Yadav S (2011) Validated HPTLC Method for Simultaneous Quantitation of Cifixime and Ofloxacin in Bulk Drug and in Pharmaceutical Formulation. Pharmacie Globale (IJCP) 2(4): 1-4.
- Bhinge SD, Malipatil SM (2016) Development and validation of a stability-indicating method for the simultaneous estimation of cefixime and dicloxacillin using the RP-HPLC method. Journal of Taibah University for Science 10(5): 734-744.
- Hussein LA, Hussein EM, Magdy N, Mohamed HS (2017) Simultaneous estimation of Ofloxacin and Cefixime in tablet form in presence of the inactive Ofloxacin USP Related compound A Bulletin of Faculty of Pharmacy, Cairo University 55(1): 171-176.
- Hassouna MEM, Abdelrahman MM, Mohamed MA (2017) Validation of a Novel and Sensitive RP-HPLC Method for Simultaneous Determination of Cefixime Trihydrate and Sodium Benzoate in Powder for Oral Suspension Dosage Form. J Forensic Sci & Criminal Inves 2(4): 555600.
- Hariprasad T, Gurumoorthy P, Ali JN (2014) Analytical Method Development and Validation of Cefixime Oral Suspension by RP-HPLC as Per ICH/USP Guidelines. IJIPBART 1(1): 47-58. 26. Patel NS, Tandel FB, Patel YD, Thakkar KB (2014) Development and Validation of Stability-indicating HPLC Method for Simultaneous Estimation of Cefixime and Linezolid. Indian J Pharm Sci 76(6): 535540.
- Rao MN, Veni PRK, Haribabu B (2015) Stability indicating rp-hplc method for determination of cefixime and ornidazole in combined pharmaceutical dosage form. RJC 8(4): 477-486.
- Patwari A, Dabhi M, Rathod I, Desai U, Suhagia B, et al. (2014) Simultaneous Determination of ofloxacin and Cefixime in combined tablet Dosage Form by HPLC and Absorption correction method. Bull Pharm Res 4(3): 112-117.
- Attimarad MV, Alnajjar AO (2013) A conventional HPLC-MS method for the simultaneous determination of ofloxacin and cefixime in plasma: Development and validation. J Basic Clin Pharma 4(2): 36-41.
- Danafar H (2015) A quick and easy high performance liquid chromatography method for evaluation of cefixime in human plasma. Pharm Biomed Res 1(4): 29-39.
- Kundu S, Majumder T, Barat PK, Ray SK (2015) Development and Validation of a HPLC-UV Method for Simultaneous Determination of Cefixime and Ofloxacin in Tablet Formulation. Int J Pharm Sci Res 6(2): 884-89.
- Elias B, Alfeen A (2016) Determination of Cefuroxime Axetil and Cefixime Trihydrate in Pharmaceutical Dosage Forms by RP-HPLC Method. Pharm Anal Chem 2: 114-118.
- Dhara P, Dhananjay M, Vandana P, Devanshi P, Hira P (2017) A Validated Stability Indicating RP-HPLC Method Development and Validation for Simultaneous Estimation of Cefixime Trihydrate and Levofloxacin Hemihydrate in Pharmaceutical Dosage Form. Int J Analytical Techn 3(1):1-12.
- Kamal MM, MD Sharif, Anisuzzaman MdS, Begum AA (2013) Comparative dissolution studies of cefixime solid dosage forms available in Bangladesh market. J Biol Sci 2(2):115-120.
- Azarmi S, Roa W, Lobenberg R (2007) Current perspectives in dissolution testing of conventional and novel dosage forms. Sci Pharm 76: 541-554.
- Mashru RC, Saikumar SV (2010) Development and validation of spectroscopic methods for simultaneous estimation and dissolution of ofloxacin and ornidazole in tablet dosage forms. Eclet Quim Sao Paulo 35(3): 123-132.
- Brown CK, Chokshi HP, Nickerson B (2004) Acceptable analytical practices for dissolution testing of poorly soluble compounds. Pharm Tech 28: 56-65.
- ICH, Q2 (R1) (2005) Validation of Analytical Procedures: Text and Methodology. ICH Harmonized Tripartite Guideline.
- Moore JW, Flanner HH (1996) Mathematical comparison of dissolution profiles. Pharm Tech 20(6): 64-74.















