



FDA 510 (k) Process- How To Get It Right The First Time?
Sunil Aggarwal* and Somya Aggarwal
Independent scholars, USA
Submission: October 13, 2016; Published: October 24, 2016
*Corresponding author: Sunil Aggarwal, Master Black Belt Lean Six Sigma, New York, USA, Tel:1-717-622-1264; Emaildrsunilaggarwal@gmail.com
How to cite this article: Sunil A, Somya A. FDA 510 (k) Process- How To Get It Right The First Time?. Canc Therapy & Oncol Int J. 2016; 2(3): 555586. DOI: 10.19080/CTOIJ.2016.02.555586
Abstract
The medical devices that are designated to be marketed need to go through a clearance process set forth by FDA. The Premarket notification (PMN) or 510(k) is the most common regulatory pathway in US but poses many challenges to medical device manufacturers. FDA has cleared more than 1, 40,000 medical devices since 1976. This is a clearance process, and not an approval, for medical devices. 510(K) submission has a purpose, a process and should be well understood in order to avoid unnecessary delays and failures.
Keywords: Medical device; Regulation; FDA; 510(K); Substantially Equivalent
Abbreviations: FDA: Food and Drug Administration, PMA: Premarket Approval; SE: Substantially Equivalent;
Introduction
Getting a clearance letter from FDA on 510(K) for a medical device is a milestone and the ultimate goal for any medical device manufacturer, be it a small or a large company. This demonstrates that the medical device so produced has demonstrated a “substantially equivalent” (SE) status to a predicate device: a medical device cleared by FDA prior to 1976. This implies that the current medical device is deemed fit to be acceptable based on the difference in the device since first clearance. Any alterations in the devices or any changes to indications, contraindications or operations require a new 510(K) submission. Lab data is almost always mandatory to be included in the submission. Reducing unnecessary waste from a system and getting most effective medical devices by quality processes such as lean is desired [2].
What is a medical device?
As per FDA:
A medical device is “an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory which is:
- Recognized in the official National Formulary, or the United States Pharmacopoeia, or any supplement to them.
- Intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or
- Intended to affect the structure or any function of the body of man or other animals, and which does not achieve any of its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of any of its primary intended purposes” [2].
Classification of Medical Devices
Based on the level of risk [3], in 1976, Medical Device Amendments to the Federal Food, Drugs and Cosmetics Act, classified all medical devices in three categories:
- Class I: Simple devices posing no or minimal risk to the user and exempt from FDA clearance, like bedpan, elastic bandages, stethoscope etc. These are about 40% of all devices manufactured.
- Class II: Moderate risk to the user and generally require FDA clearance before getting marketed, like sutures, condoms, X-Ray systems etc. Some are FDA exempt and some require a PMA. These are about 50% of all devices manufactured [4,5].
- Class III: These life- supporting or life-sustaining devices pose a serious risk to the user and require a PMA and FDA clearance before marketed like, stents, implants, heart pumps etc. These require a PMA, as well [6-9].
What is PMA?
Premarket Approval (PMA) is the demonstration by the medical device manufacturer of the safety of the intended use and the usefulness of a new medical device to the FDA, of which FDA has no prior knowledge or experience. This would always include human and lab trial data. Class III devices always require a PMA and other such devices that do not qualify under 510(k) submission.
What are 510 (k)?
It is section 510-360 subsection k-Registration of producers of drugs or devices. As per this submission, the producer of the medical device must demonstrate that this medical device is substantially equivalent to a predicate device that has been cleared previously by FDA. There is a clearance given by FDA and not an approval. If there is a change in the use, contraindication or operation of use as compared to a previous cleared medical device, then this new device would require a 510(k) submission.
K number from FDA
All medical Devices submitted for 510(K), at least ninety days before introduction of a device, are assigned a K number followed by six digits. These six digits have significance to it: the first two digits being the year of submission and the last four digits begin with a zero in the order it was received for a review. Zimmer was the one which has 50(K) number K760001 indicating it to be the first submission since FDA in 1976. Boston Scientific Scimed Inc. has K0000, being having this number allotted in a new millennium on January 3, 2000.
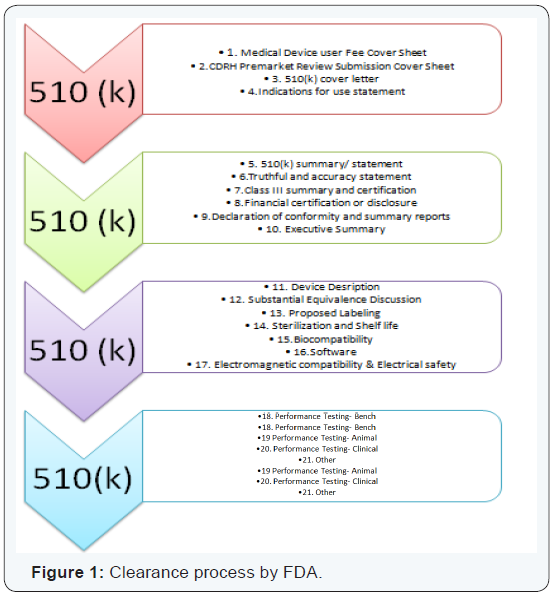
How to format a 510(k)?
A 510(k) submission, not only requires a sound understanding of the process of submission, but also requires a thorough formatting with substantial inclusions. Some of the material that needs to be included is:
- ( h t t p : / / w w w . f d a . g o v / M e d i c a l D e v i c e s / DeviceRegulationandGuidance/GuidanceDocuments/ucm084365. htm) (Figure1).
Conclusion
FDA is there to safeguard the interest of the public and make sure the efficacy and safety of a medical device [5,8]. The classification of a medical device, predicate device, premarket approval, and 510 (k) are important concepts in regulation of a medical device. The new medical device should be innovative and support public health. Some people argue that 510(k) is inefficient and time consuming for newer devices to come into the market and some also have doubts on the ability of 510(k) on the safety and efficacy of a medical device. Some companies have been seen to defraud FDA on the medical devices. Whatever it may be, 510(k) is a process in place that may be eventually see more improvements [1,4].
References
- Pietzsch JB, Zanchi MG, Linehan JH (2012) Medical Device Innovators and the 510(k) Regulatory Pathway: Implications of a Survey-Based Assessment of Industry Experience. ASME. J Med Devices 6(2): 021015.
- Sunil A, Sanju A, Somya A (2016) A Study of Lean in Oncology: Reducing Waste and Increasing Value. Canc Therapy & Oncol Int J 1(3): 555563.
- Pietzsch JB, Aquino LM, Yock PG, Paté-Cornell M, Linehan JH (2007) Review of U.S. Medical Device Regulation.ASME. J Med Devices 1(4): 283-292.
- Daniel BK, ShuaiXu, Aaron SK (2012) How Does Medical Device Regulation Perform in the United States and the European Union? A Systematic Review. PLoS Med 9(7): 1001276.
- Maisel WH (2008) Semper fidelis-consumer protection for patients with implanted medical devices. N Engl J Med 358: 985-987.
- Sapirstein W, Alpert S, Callahan TJ (1994) The role of clinical trials in the Food and Drug Administration approval process for cardiovascular devices. Circulation 89(4): 1900-1902.
- US Food and Drug Administration, Medical devices premarket approval (PMA). Accessed November 9, 2010.
- Public health effectiveness of the FDA 510(k) clearance process: balancing patient safety and innovation: workshop report (2010).
- Maisel WH (2009) Medical devices: are current regulations doing enough for patients? testimony before the Subcommittee on Health, Committee on Energy and Commerce, House of Representatives.
