



Brachial Plexus Neurofibroma Treated with Volumetrically Modulated Arc Therapy (VMAT): A Case Report.
Luis Moreno Sánchez1*, Jacksson Sánchez1 and Moisés Dieguez2
1Department of Radiotherapy, National Cancer Institute Rosa Emilia Sánchez P. de Tavares (INCART), Dominican Republic
2CDD Radioterapy, Abreu Clinic, Dominican Republic
Submission: October 05, 2016; Published: October 24, 2016
*Corresponding author: Luis Moreno Sánchez. Radiation oncologist, Intra and Extracranial radiosurgery, brachytherapy, Coordinador de Radioncología, Instituto Nacional del Cáncer Rosa de Tavares (INCART), C/Correa y Cidrón, esq. Rafael Sánchez Ravelo. Zona Universitaria, Santo Domingo, Dominican Republic
How to cite this article: Luis M S, Jacksson S, Moisés D. Brachial Plexus Neurofibroma Treated with Volumetrically Modulated Arc Therapy (VMAT): A Case Report.. Canc Therapy & Oncol Int J. 2016; 2(2): 555585. DOI: 10.19080/CTOIJ.2016.02.555585
Abstract
Neurofibromatosis was first described in 1882 by Friedrich Daniel von Reckling hausen, a German pathologist. Neurofibroma is a benign peripheral nerve sheath tumor that consists of Schwann cells, associated or unassociated with axons, perineural cells, and fibroblasts. Whenever possible, the treatment of choice should be surgical, but the management depends on the location and growth pattern. We present the case of a patient with left axillary neurofibroma without neurofibromatosis (NF) in whom surgery was delayed due to involvement of the brachial plexus, so was sent to radiotherapy, planned and treated with volumetrically modulated arc therapy (VMAT).
Keywords: Neurofibroma; VMAT; Radiotherapy; Axillary Tumor; INCART
Abbreviations: NF: Neurofibromatosis; VMAT: Volumetrically Modulated Arc Therapy; CRO: Radiation Oncology Center; INCART: Institute Rosa Emilia Sánchez Pérez de Tavares; CW: Clockwise; NTO: Normal Tissue Objective; OAR: Organ at Risk
Introduction
Neurofibromas are benign tumors that arise from nerves and are composed of Schwann cells, perineural cells, and fibroblasts [1]; typically arise in the setting of neurofibromatosis type I (NF I) [2], but may also occur independently, when it comes to multiple tumors is the main clinical manifestation of the different types of neurofibromatosis and are processes that are transmitted with autosomal dominant inheritance with variable penetrance. In most cases are treated by surgery, especially when they are symptomatic or with evident anatomical changes [3,4], but when a neurofibroma involves a particularly long segment of nerve or nerves, are generally impossible to remove without removing the entire nerve, causing a major neurological deficit, therefore, these variants are not usually subjected to surgery. In this report, we describe an axillar neurofibroma in a patient without NF I, which was treated with radiotherapy using volumetrically modulated arc therapy (VMAT).
Case Summary
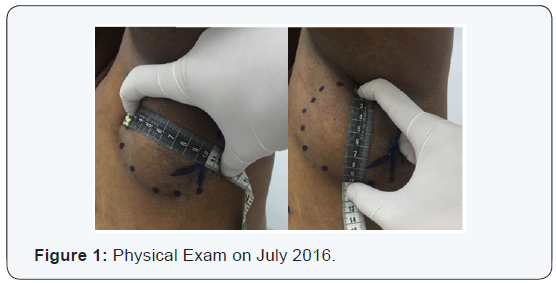
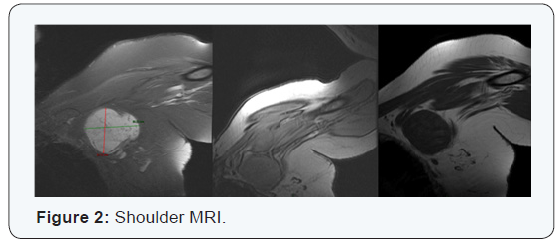
Female patient, 55 years old, who had previously presented to an external surgery department was remitted to the Radiation Oncology Center (CRO) at National Cancer Institute Rosa Emilia Sánchez Pérez de Tavares (INCART), in Santo Domingo, Dominican Republic, with history of pain and volume increase in left armpit, diagnosed as non-surgical axillar neurofibroma. Medical record, including MRI, CT and pathologic specimen, were reviewed. The patient was considered initially for surgery like standard treatment, but during process evidenced brachial plexus commitment, because of this was omitted the surgery and was referred to radiation oncology. The Physical exam reported: Karnofsky (KPS) 100%, ECOG 0, in the left axillary region was felt a mass about 10 cm in diameter (Figure 1), mobile, painful, hard consistency with left arm weakness. A shoulder MRI with gadolinium reported: oval image 8.5 x 5.3 cm anteroposterior, anterior to the subclavian neurovascular bundle located in the anatomical path of the brachial plexus, highly suggestive of neural image type neurofibroma. Described image shows a bottom center 1.9 cm cystic degeneration, in turn shows an intense post gadolinium enhancement and heterogeneous accordance with important vascularization (Figure 2).
Simulation Ct and delineation
The patient was positioned in supine using a table for breast carbon fiber of the CIVCO® brand. Package tumor localization (TumorLOC®) Brilliance CT was used for the location of tumor tomography simulation. This tool allows you to place the isocenter manually depending on the selected organs and can create orthogonal beams associated with that default position of isocenter. Tomographic scanning technique was 120kV, axial cut, over 200,3 mm apart.
Field arrangement and treatment plan
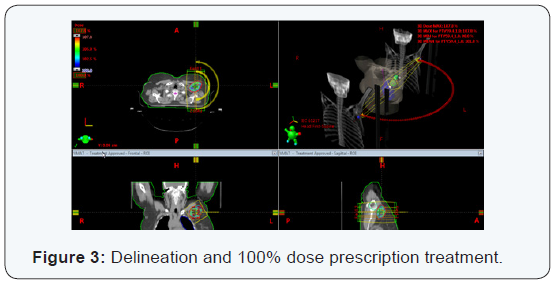
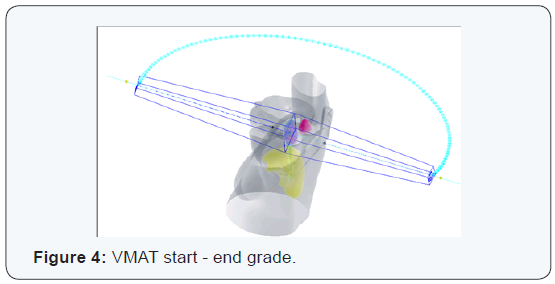
The treatment technique corresponded to volumetrically modulated arc therapy (VMAT) for this, two semi-arches were used. The first from 0° to 179° towards the direction of clockwise (CW) and the second reverse direction arc (179° to 0°). They were established for each arc 98 control points with maximum speed of gantry 4.8 deg / s. Each semi-arch lasted 48 seconds (0.8min) (Figure 3 & 4). The bodies’ risks were considered for planning spinal cord, left lung, left humeral head and left mammary gland. As establishment of an NTO (normal tissue objective) use the automatic option. Resolutions (mm) and priorities (%) planning for OAR (organ at risk) were, respectively: spinal cord 2 mm and 35%, humeral head 2.65 mm and 25%, left lung 3 mm and 30%, left breast 3 mm and 25%.
Assessment treatment plan
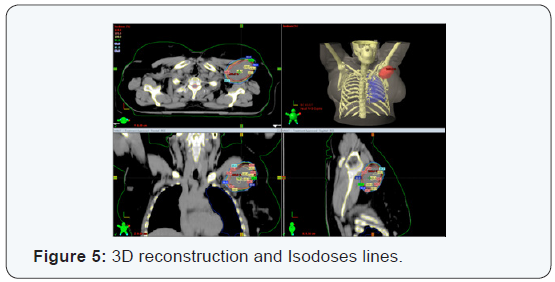
Evaluating different plane cuts ensure that 100% of the prescribe dose is within the volume of total dose planning 5940 cGy. The bodies assessed risks are below the limits set by Quantec / RTOG (Figure 5).
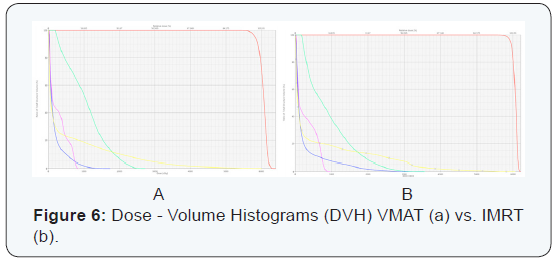
One of the benefits of this proposal lies among other things in the runtime patient treatment compared to some other CRT3D or IMRT technique. The maxim and average doses to OAR were: 27.3 Gy / 10.5 Gy (VMAT) and 36 Gy / 9.12 Gy (IMRT)for left humeral head (green); 60.0 Gy / 5.2 Gy (VMAT) and 60.5 Gy / 5.7 Gy (IMRT) for left breast (yellow); 8.5 Gy / 2.7 Gy (VMAT) and 9.6 Gy / 2.9 Gy (IMRT)for spinal cord(purple) and finally 17.3 Gy / 1.77 Gy (VMAT) and 35.5 Gy / 2.5 Gy (IMRT) for left lung (blue) (Figure 6)
QA
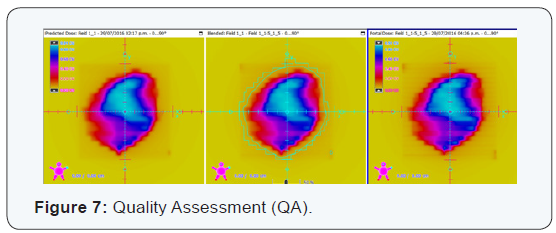
The two semi-arches were evaluated for creep refers using an amorphous silicon system to perform dosimetry portal. Descriptors parameters for evaluation were the index gamma 3% and distance agreement (DTA) 3mm. Predicted doses vs. the portals doses obtained successfully passed the evaluation criteria (Figure 7).
Diagnosis
Left axillary neurofibroma. The microscopic biopsy description reported: sections showed mesenchymal neoplasm, partially encapsulated, constituted by the proliferation of spindle cells, elongated nuclei arranged in swirling pattern and fibrillar collagenous stroma storiform on congestive vessels with thickened walls and areas of bleeding. Differential diagnoses included: schwannoma, malignant peripheral nerve sheath tumor and desmoid tumor [5].
Discussion
Neurofibromatosis was first described in 1882 by Friedrich Daniel von Recklinghausen, a German pathologist. It is a disease caused by an abnormality in a gene on chromosome 17.Thediagnosis of Neurofibromatosis type I (NF1) is confirmed If 2 or more of these criteria are met in an individual: six or more stains coffee with milk greater than 5 mm in pre pubertal patients and over 15 mm in post pubertal patients; two or more neurofibromas of any type or one plexiform neurofibroma; sign Crowe (axillary or inguinal freckling); [6-9] Glioma optic nerve; two or more Lisch nodules (iris harmartomas); typical bone lesions (dysplasia of the sphenoid dysplasia or thinning of long bone cortex with or without pseudoarthrosis); history of neurofibromatosis type I in parents or siblings [10]. Meanwhile, to make the diagnosis of NF 2 is necessary to have the following criteria: bilateral masses in the eighth cranial demonstrated techniques by appropriate images or first-degree relative with NF2 and one of the following: unilateral mass the eighth cranial nerve, or two of the following: neurofibroma, meningioma, glioma, schwannoma, subcapsular lenticular opacity youth back [10].
In the case of our patient, it has none of the criteria for NF1or 2. Neurofibroma is a benign peripheral nerve sheath tumor that consists of Schwann cells, associated or unassociated with axons, perineural cells, and fibroblasts. The tumor routinely expresses S-100 protein. Benign neurofibromas most often occur in association with neurofibromatosis type 1 (NF1) but may also occur sporadically [1,2], like in our case report.
The biological behavior of neurofibromas is usually benign, but it is not rare sarcomatous degeneration in plexiform. Neurofibromas occur in multiple locations, exhibit different growth patterns and morphologies at highly variable rates, a classification system stratifies these tumors into 5 types: localized cutaneous, diffuse cutaneous, localized intraneural, plexiform, and massive soft tissue neurofibromas [4].
The management depends on the location and growth pattern. Whenever possible, the treatment of choice should be surgical, especially when they produce functional or cosmetic déficits, ideally with complete removal of the tumor, but some tumors diffusely involve adjacent normal tissues such as nerves and blood vessels, making complete remove of the tumoral most impossible [1,3,4,6,8,9]. Medical therapies have been investigated, most are still investigational [4] and Radiotherapy has been rarely used like initial treatment, but by default in patients with incomplete resection [1,4]. Needle et al. [6] reported 121 children with plexiform neurofibromas who underwent resection and were followed for over 20 years. Those that were entirely removed were less likely to recur and had a longer median time to failure. Near-total and subtotal resections demonstrated recurrence rates of 39.5% and 44%, respectively.
Conclusion
We must emphasize team work and multi disciplinary approach to tackling the disease. The commitment caused by neurofibromas depends in much of their location, skin lesions usually cause deformity, while more injuries shallow tend to generate functional compromise. Despite being mostly benign, some cause destruction secondary to the pressure exerted, so the symptoms are depend on the size of the lesion. The therapeutic approach should be multidisciplinary, with the initial treatment recommended surgery, but in case of not being able to perform or partial resection, radiotherapy with doses between 54-60 Gy can achieve adequate local control. In reviewing the literature an extensive description of the disease and epidemiological data was found are scarce, so studies to know the disease more accurately are needed.
References
- Robertson TC, Buck DA, Schmidt-Ullrich R, Powers CN, Reiter ER (2004) Isolated Plexiform Neurofibroma: Treatment with Three-Dimensional Conformal Radiotherapy. Laryngoscope 114(7): 1139-1142.
- Riccardi VM (1991) Neurofibromatosis: Past, present, and future. N Engl J Med 324(18): 1283-1285.
- Mehta D, Mehta D, Shaam M (2013) Huge Axillary Mass - Neurofibroma Brachial Plexus. Indian Journal of Surgery 75(1): S47-S50.
- Chopra R, Morris CG, Friedman WA and Mendenhall WM (2005) Radiotherapy and Radiosurgery for Benign Neurofibromas. Am J Clin Oncol 28(3): 317-320.
- Binder DK, Smith JS, Barbaro NM (2004) Primary brachial plexus tumors: imaging, surgical, and pathological findings in 25 patients. Neurosurg Focus 16(5): 1-6.
- Needle MN, Cnaan A, Dattilo J, Chatten J, Phillips PC, et al. (1997) Prognostic signs in the surgical management of plexiform neurofibromas: the Children’s Hospital of Philadelphia experience, 1974 -1994. J Pediatr 131(5): 678 - 682.
- Scheithauer B, Woodruff J, Erlandson R (1999) Tumors of the peripheral nervous system. In: Rosai JR, Sobin LH, (Eds.) Atlas of Tumor Pathology, (3rd edn), Armed Forces Institute of Pathology, Washington, DC, USA.
- Kim DH, Murovic JA, Tiel RL, Moes G, Kline DG (2005) A series of 397 peripheral neural sheath tumors: 30-year experience at Louisiana State University Health Sciences Center. J Neurosurg 102(2): 246-255.
- Lusk MD, Kline DG, Garcia CA (1987) Tumors of the brachialplexus.Neurosurgery 21(4): 439-453.
- Neurofibromatosis (1988) Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 45(3): 575-578.
