



Pleural Malignant Solitary Fibrous Tumor Mimicking a Nerve Sheath Tumor in a Patient with Neurofibromatosis Type 1
William Green*, Michael Sherman, Huijian Sun, Ranjana Nawgiri, Nahal Boroumand and Luba Frank
University of Texas Medical Branch, USA
Submission: October 15, 2016; Published: October 19, 2016
*Corresponding author: William Green, University of Texas Medical Branch, 301 University Blvd, Galveston, TX 77555, USA.
How to cite this article: Green W, Sherman M, Sun H, Nawgiri R, Boroumand N, Frank L. Pleural Malignant Solitary Fibrous Tumor Mimicking a Nerve Sheath Tumor in a Patient with Neurofibromatosis . Canc Therapy & Oncol Int J. 2016; 2(1): 555581. DOI:10.19080/CTOIJ.2016.02.555581
Abstract
Neurofibromatosis type 1 (NF1), formerly known as von Recklinghausen’s disease is a nervous system tumor disorder of variable expressivity. Malignant solitary fibrous tumors (SFT) are rare spindle cell soft tissue tumors. We report a case of a 35-year-old male who presented clinically with NF1 and a mass that mimicked a nerve sheath tumor radiologically, but pathologically was, in fact, a malignant solitary fibrous tumor. Based on a literature review, the association between NF1 and malignant SFT has never been published.
Keywords: Neurofibromatosis type 1; Malignant solitary fibrous tumor
Abbrevations: SFT: Solitary Fibrous Tumors; NF1: Neurofibromatosis type 1
Mini Review
Neurofibromatosis type 1 (NF1) is a multisystem disease characterized by cutaneous findings, as well as benign and malignant tumors of the nervous system. NF1 occurs in about 1 in 3,000 individuals, and about half of the affected individuals are first cases in their family [1].
A solitary fibrous tumor (SFT) is a rare spindle cell tumor that most commonly arises in the pleura. These tumors comprise only 1-2% of all soft tissue tumors, and of those, between 12 to 22% are malignant [2-3].
No link has ever been made between NF1 and a malignant SFT Conzo et al. [4-5] first described SFT in an NF1 patient, and Yamamoto, et al later reported another case, but neither case dealt with malignant SFT.
Here, we describe a patient with clinical NF1, who presented with a mass that mimicked a nerve sheath tumor, but biopsy and immunohistochemical assays showed that the correct diagnosis was of a malignant solitary fibrous tumor.
Case Presentation
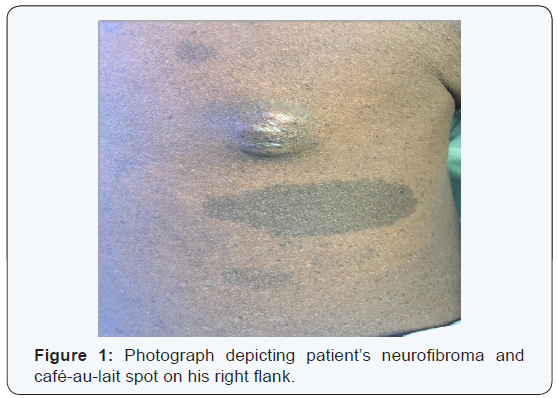
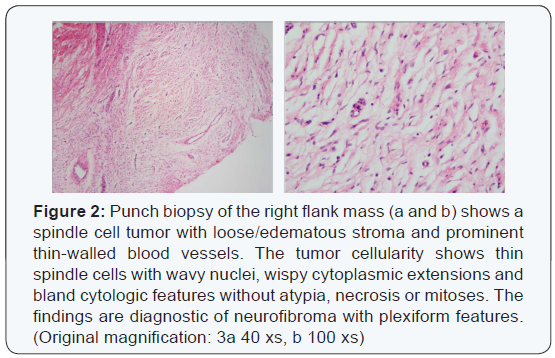
In August 2015, a 35-year-old man presented from an outside hospital with chest pain, recurrent left lower lobe pneumonia and a pleural mass seen on chest radiograph. Physical exam revealed multiple café-au-lait spots on his face, back, chest, abdomen, and legs that ranged from 5mm to 15mm, as well as nodules present on the patient’s right flank, as seen in (Figure 1), and lateral to his left eyebrow. Ophthalmologic exam revealed bilateral Lisch nodules. Due to the array of clinical findings, a biopsy of the right flank nodule was performed and revealed a neurofibroma with plexiform features (Figure 2).
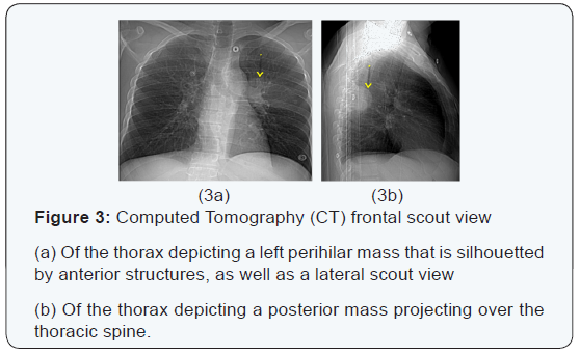
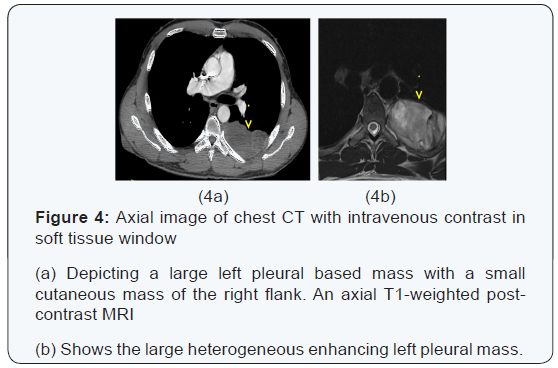
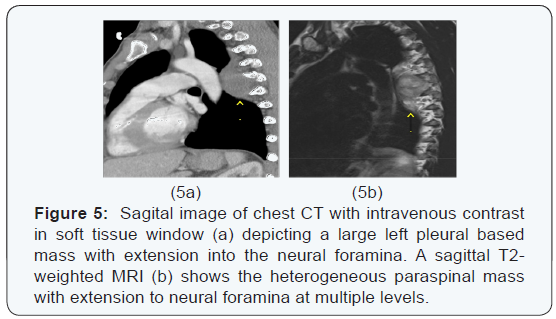
The chest x-ray from the outside hospital was not available at the time of writing, but, as a surrogate, scout images from the initial CT scan performed in our institution may be seen in (Figure 3), displaying the thoracic mass initially appreciated at the outside hospital. A CT scan of the thorax with contrast was performed in our institution, which showed a left posterior intrathoracic soft tissue mass with areas of enhancement, neural foraminal widening and rib splaying (Figures 4a & 5a). An MRI of the same area was performed (Figure 4b & 5b) to better show the mass in question, and a biopsy was recommended as the mass was more suggestive of a nerve sheath tumor, or, less likely, a pleural fibroma.
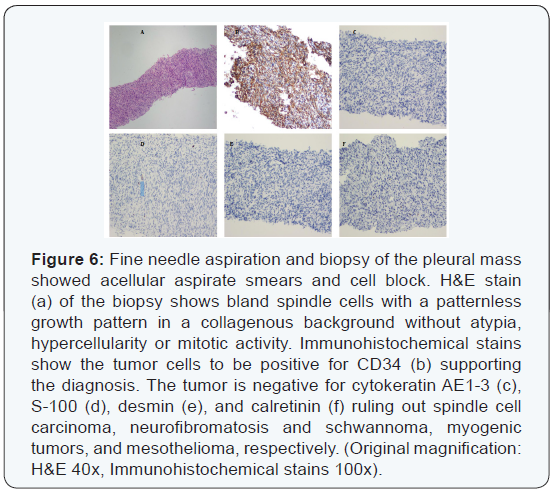
Ultrasound-guided fine needle aspiration and biopsy of the left pleural mass showed bland spindle cells without defined architectural pattern within a collagenous background without necrosis or mitotic activity. Immunohistochemical staining was positive for CD34, supporting a diagnosis of solitary fibrous tumor (Figure 6).
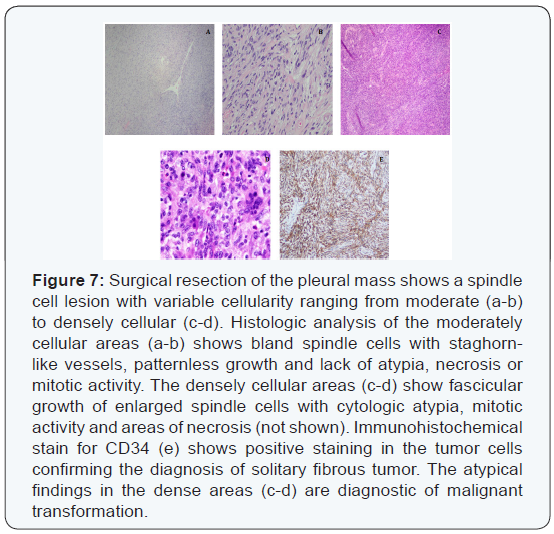
Subsequently, the pleural mass was resected and histologic evaluation showed a spindle cell lesion with biphasic morphology. The majority of the tumor showed classic solitary fibrous tumor with benign features (Figures 7a & b) with a portion of the tumor showing markedly increased cellularity, cytologic atypia and increased mitotic activity (>4/10 high power fields) diagnostic of malignant transformation (Figures 7c & d). Immunohistochemical stain for CD34 (Figure 7e) was positive, confirming the findings.
Discussion
Neurofibromatosis type 1 (NF1), also known as von Recklinghausen’s disease, is caused by an alteration of the NF1 gene, which is a tumor suppressor gene located on the long arm of chromosome 17 (17q11.2). While genetic testing is not required, it is helpful in confirming the diagnosis. The NF1 gene encodes a protein called neurofibromin, which is a member of the GTPase activating protein family. A loss of function mutation to the NF1 gene results in increased cellular proliferation that manifests as multisystemic tumorous growth. Neurofibromatosis can be diagnosed clinically if at least 2 of the 7 following criteria are fulfilled: 6 or more hyperpigmented macules, axillary or inguinal freckles, two or more neurofibromas or one plexiform neurofibroma, optic nerve glioma, two or more iris hamartomas, body dysplasia, or a first degree relative with NF1 [6].
On the other hand, SFT classically presents as a pleural mass. Klemperer and Rabin first described the tumor in the pleura in 1931 [7]. Most of the tumors are benign neoplasms that are diagnosed in adults with an age range between 50-70 years-old and a male/female ratio of approximately 1 [8-10]. Of the pleural-based SFTs, most arise from visceral pleura (80%) with the remainder arising from parietal pleura [9]. Gross examination of an SFT typically shows a smooth glistening capsule surface [9]. Histologically, most tumors are composed of collagen-forming spindle-cells, arranged in interlacing fascicles with a classic “patternless” pattern [2]. The morphologic findings are typical, though not always demonstrable in small biopsy sections; therefore the characteristic immunohistochemical positivity for CD34 is significantly helpful in separating SFT from other spindle cell neoplasms [11]. Criteria for malignancy include the presence of necrosis, increased mitotic activity, greater cellularity, and cellular polymorphism [10]. Malignant SFT is diagnosed with presence of at least one of these criteria [10]. Approximately 15-35% of SFT are malignant at the time of diagnosis with clinical characteristics of higher recurrences and lower overall survival [2]. Nonetheless, regardless of benign.
Or malignant presentation of SFT, the hallmark of treatment is large en-bloc resection of the primary tumor with close clinical surveillance for recurrences. Mediastinal lymph node dissection and adjuvant treatments such as radiotherapy and chemotherapy are not usually employed for treatment purposes, but rather as palliative options for cases of distant metastasis and locally advanced recurrence that are not amenable to surgery [2].
Historically, NF1 has been associated with various tumors, most notable of which are malignant peripheral nerve sheath tumor, rhabdomyosarcoma, and gastrointestinal stromal tumor (GIST), with an overall occurrence rate that reaches up to 5-13%, 0.5%, and 6% respectively [12-14]. However, there are no systematic reviews that link NF1 with development of SFT. Only two case reports have been published with anecdotal evidences where SFT were found concurrently in NF1 patients. Of these two reports, both patients had pathologic diagnoses of benign SFTs. Specifically, Conzo et all described a patient who had an encapsulated tumor arising from the upper pole of the right kidney that mimicked an adrenal gland or renal tumor, but later proved to be a SFT [4]. Yamamoto et al described a patient who underwent Tc-99m MIBI parathyroid scinitigraphy, which showed uptake highly suggestive of an ectopic parathyroid adenoma, but also later proved to be a SFT [5]. Our case appears to be the first documented case of malignant pleural SFT in an NF1 patient, which also showed signs of mimicry, as seen in the first two cases. Initial radiologic evaluation of the pleural mass suggested nerve sheath tumor, especially in the setting of other clinical findings of neurofibromatosis such as plexiform neurofibroma, as well as presence of a para-orbital mass. Nonetheless, other neoplastic causes such as pleural lipoma, pleural fibrosarcoma, mesothelioma, and metastatic pleural disease could not be ruled out by imaging alone without a definitive biopsy of the mass. Therefore, fine needle aspiration of the mass was performed and showed diagnostic features of SFT and subsequent excision showed areas of malignant transformation.
While NF1 patients have an increased risk of developing various tumors and malignancies, there is no definitive link between NF1 and SFT due to lack of published data [15]. Hence, it is difficult to comment on the exact relationship between these two disease processes. Whether or not NF1 is associated with the development of SFT and the relationship between such, as well as possible malignant transformation of SFT would need to be investigated further.
References
- Friedman JM (1998) Neurofibromatosis 1. In: Pagon RA, Adam MP, Ardinger HH, GeneReviews, University of Washington, Seattle, USA.
- Penel N, Amela EY, Decanter G, Robin YM, Marec-Berard P (2012) Solitary fibrous tumors and so-called hemangiopericytoma. Sarcoma 2012: 690251.
- Robinson LA (2006) Solitary fibrous tumor of the pleura. Cancer Control 13: 264-269.
- Conzo G, Tartaglia E, Gambardella C (2014) Suprarenal solitary fibrous tumor associated with a NF1 gene mutation mimicking a kidney neoplasm: implications for surgical management. World Journal of Surgical Oncology 12(1): 87.
- Yoko Yamamoto, Ken Kodama, Shigekazu Yokoyama, Masashi Takeda, Shintaro Michishita (2015) “A Pleural Solitary Fibrous Tumor, Multiple Gastrointestinal Stromal Tumors, Moyamoya Disease, and Hyperparathyroidism in a Patient Associated with NF1,” Case Reports in Surgery 2015: 5
- Ferner RE, Huson SM, Thomas N (2007) Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. Journal of Medical Genetics 44(2): 81-88.
- Klemperer P, Rabin CB (1931) Primary neoplasms of the pleura: a report of five cases. Arch Pathol 11: 385-412.
- Cardillo, L Carbone, F Carleo (2009) “Solitary fibrous tumors of the pleura: an analysis of 110 patients treated in a single institution.” The Annals of Thoracic Surgery 88(5): 1632-1637.
- P Magdeleinat, M Alifano, A Petino (2002) “Solitary fibrous tumors of the pleura: clinical characteristics, surgical treatment and outcome.” European Journal of Cardio-Thoracic Surgery 21(6): 1087-1093.
- DM England, L Hochholzer, M J McCarthy (1989) “Localized benign and malignant fibrous tumors of the pleura. A clinicopathologic review of 223 cases.” American Journal of Surgical Pathology 13(8): 640-658.
- Han Y, Zhang Q, Yu X (2015) Immunohistochemical detection of STAT6, CD34, CD99 and BCL-2 for diagnosing solitary fibrous tumors/hemangiopericytomas. International Journal of Clinical and Experimental Pathology 8(10): 13166-13175.
- Miettinen, Fetsch JF (2006) “Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetics study of 45 cases.” The American journal of surgical pathology 30(1): 90-96.
- MJ McMaster, EH Soule, JC Ivins (1975) “Haemangiopericytoma: a clinico-pathological study and long term follow-up of 60 patients.” Cancer 36(6): 2232-2244.
- McGaughran JM, Harris DI, Donnai D, Teare D, MacLeod R, et al. (1999) A clinical study of type 1 neurofibromatosis in north west England. J Med Genet 36(3): 197-203.
- Sung L, Anderson JR, Arndt C, Raney RB, Meyer WH, et al. (2004) Neurofibromatosis in children with Rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma study IV. J Pediatr 144(5): 666-668.
- Ruggieri M, Huson SM (2001) The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology 56(11): 1433-1443.
