




In Silico Analysis of Single Nucleotide Polymorphisms (SNPS) in Human Abetalipoprotein Epsilon 4 (APOE E4) Gene as a Cause of Alzheimer's Disease from Genetic Mutation to Functional Predication from Structural Change Patterns
Abdelmohymin AA Abdalla*, Mohamed Elsheikh, Ahmed Babiker, Mohanad Abdelrahim, Mohamed Abdelrahim, Mohamed Taha, Mohamed Muneer, Mahil Abdalla, Mohamedahamed Hassan and Abbashar Hussein
Department of Bioinformatics, University of Khartoum, Sudan
Submission: March 28, 2017; Published: May 30, 2017
*Corresponding author: Abdelmohymin AA Abdalla, Department of Bioinformatics, University of Khartoum, Al-Gamaa Ave, Khartoum 11111, Sudan, Email: shawrma214@gmail.com
How to cite this article: Abdelmohymin A A, Mohamed E, Ahmed B, Mohanad A, Mohamed A et al. In Silico Analysis of Single Nucleotide Polymorphisms (SNPS) in Human Abetalipoprotein Epsilon 4 (APOE E4) Gene as a Cause of Alzheimer�s Disease from Genetic Mutation to Functional Predication from Structural Change Patterns. Curr Trends Biomedical Eng & Biosci. 2017; 5(1): 555653. DOI: 10.19080/CTBEB.2017.05.555653
Abstract
Alzheimer disease (AD) is an acquired disorder of cognitive and behavioral impairment that markedly interferes with social and occupational functioning. Single-nucleotide polymorphisms (SNPs) play a major role in the understanding of the genetic basis of this complex disease. In this work, we have analyzed the genetic variation that can alter the expression and the function of the APOE e4 gene by analysing the SNPs in the coding regions of APOE e4 gene using computational methods. Genomic analysis of APOE e4 was initiated Polyphen and SIFT server used to retrieve 10 harmful mutations, among of these nsSNPs damaged SNPs six non-synonymous SNPs showed very damaging by higher PSIC score of the Polyphen server with a SIFT tolerance index of 0.00-0.01 (R50C, L115S, R132S, E139V, E150G, R168H); and one in very highly conserve region (R154P). Screening for these SNPs variants in coding region may help in Alzheimer disease molecular and genetic early diagnosis.
Analysis Of the total 6 SNPs in 3'UTR region of APOE e4 gene by its special tool PolymiRTS show all the 3 UTR containing alleles insignificant to make any distribution in conserve miRNA site. We also predict the function of related genes and gene sets by using Gene MANIA which finds other genes that are related to APOE genes, using a very large set of functional association data.
Keywords: SNPs; Apo e4; Alzheimer disease; In silico analysis
Abbreviations: AD: Alzheimer Disease; SNPs: Single-Nucleotide Polymorphisms; NFTs: Neurofibrillary Tangles; CTFs: C-Terminal Fragments; ESNP: Ensembles Protein; AAS: Amino Acid Substitution; SIFT: Sorting Intolerant From Tolerant
Introduction
Alzheimer's disease (AD) is the most common cause of dementia in the elderly [1]. Dementia is an acquired deterioration in cognitive abilities that impairs the successful performance of activities of daily living [2]. Memory is the most common cognitive ability lost with dementia [3]. Pathologically, atrophy is distributed throughout the medial temporal lobes, as well as lateral and medial parietal lobes and lateral frontal cortex. Microscopically, there are neurotic plaques containing neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau filaments, and accumulation of amyloid in blood vessel walls in cortex and lepto-meninges [4]. The identification of four different susceptibility genes for AD: APOE, APP, PSEN1, and PSEN2. By far the most commonly and most consistently associated gene mutation is in the 19 chromosome in epsilon 4 of APOE gene [5].
The major constituent of these plaques is the neurotoxic amyloid-beta-APP 40-42 peptide (s), derived proteolytically from the transmembrane precursor protein APP by sequential secretase processing. The cytotoxic C-terminal fragments (CTFs) and the caspase-cleaved products such as C31 derived from APP, are also implicated in neuronal death [6]. The APOE4 allele is genetically associated with the common late onset familial and sporadic forms of Alzheimer disease [7-11]. The presence of the APOE4 allele was strongly associated with reduced CSF levels of beta-amyloid-42. The findings suggested an involvement of ApoE in beta-amyloid metabolism. The APOE was found to be located in 19q13.20 chromosome with GeneID 348. It was found to be associated with 75 diseases including Alzheimer's disease, Hyperlipoproteinemia and Diabetic Retinopathy.
The presence of the E4 allele was associated with greater brain atrophy on imaging studies, AD E4 allele carriers showed greater atrophy in the bilateral parietal cortex and right hippocampus. It was found an association between presence of the E4 allele and the typical amnestic phenotype, characterized by initial presentation of forgetfulness and difficulties with memory. Those with the memory phenotype were 3 times more likely to carry an E4 allele compared to AD patients who displayed a non-memory phenotype. Risk for AD increased from 20% to 90% and mean age at onset decreased from 84 to 68 years with increasing number of APOE4 alleles. Thus APOE4 gene dose is a MAJOR RISK FACTOR for late onset AD and, in these families, homozygosity for APOE4 was virtually sufficient to cause AD by age 80 [3].
The mechanism by which APOE4 participates in pathogenesis is not known .Mediates the binding, internalization, and catabolism of lipoprotein particles. It can serve as a ligand for the LDL (Apo B/E) receptor and for the specific Apo-E receptor (chylomicron remnant) of hepatic tissues [12].
Materials and Methods
The critical step in this work was to select SNPs for analysis by computational softwares ; the selection was prioritizing SNPs in the coding region (axonal SNPs) that are non-synonymous (nsSNP) and SNPs at untranslated region at 3’ends (3'UTR) to predict the effect on miRNA binding on these regions that may greatly associated with tumour progression. The SNPs and the related ensembles protein (ESNP) were obtained from the SNP database (dbSNPs) for computational analyse from http://www. ncbi.nlm.nih.gov/snp/ [13] and Uniprot database http://www. uniprot.org [14] for related protein sequences.
Predicting damaging amino acid substitutions using SIFT: Updated versions of Ensemble gene annotation (GRCh37 Ensembl 66) and NCBI dbSNP database (Build 137)
SIFT is an online computational tool to detect a harmful non- synonymous single-base nucleotide polymorphism (nsSNP); SIFT predicts whether an amino acid substitution affects protein function. SIFT prediction is based on the degree of conservation of amino acid residues in sequence alignments derived from closely related sequences, collected through PSI-BLAST. SIFT can be applied to naturally occurring non-synonymous polymorphisms or laboratory induced missense mutations the genetic mutation that causes a single amino acid substitution (AAS) in a protein sequence subsequently altering the carrier's phenotype and health status. The software traces AAS and Sorting Intolerant from Tolerant (SIFT) and predicts whether these substitutions affect protein function by usingsequence homology, SIFT predicts the effects of all possible substitutions at each position in the protein sequence. Furthermore, the algorithm performs a comprehensive search in protein repositories to find the tolerance of each candidate compared to the conserved counterparts. Non-synonymous reference SNPs identity (rsSNPs ID) were downloaded from online dbSNPs of NCBI, and then submitted to SIFT. Results are expressed as damaging (deleterious) or benign (Tolerated) depending on cutoff value 0.05; as values below or equal to 0.0-0.04 predicted to be damaging or intolerant while (0.05_1) is benign or tolerated, then the damaging SNPs were re-analysed by Polyphen software which predicts the effect of mutations on both structural and functional sides. SIFT is available as online tool at http://sift. jcvi.org [15]
Prediction offunctional modification using Polyphen-2 (Polymorphism Phenotyping v2)
It is a software tool to predict possible impact of an amino acid substitution on both structure and function of a human protein by analysis of multiple sequence alignment and protein 3D structure, in addition it calculates position-specific independent count scores (PSIC) for each of two variants, and then calculates the PSIC scores difference between two variants. The higher a PSIC score difference, the higher the functional impact a particular amino acid substitution is likely to have. Prediction outcomes could be classified as probably damaging, possibly damaging or benign according to the value of PSIC as it ranges from (0_1); values closer to zero considered benign while values closer to 1 considered probably damaging and also it can be indicated by a vertical black marker inside a colour gradient bar, where green is benign and red is damaging. nsSNPs that predicted to be intolerant by Sift has been submitted to Polyphen as protein sequence in FASTA format that obtained from UniproktB /Expasy after submitting the relevant ensemble protein (ESNP) there, and then we entered position of mutation, native amino acid and the new substituent for both structural and functional predictions. PolyPhen version 2.2.2 is available at http://genetics.bwh.harvard.edu/pph2/index.shtml [16]
Protein modelling
Project hope (version 1.0): Project hope is an online webserver to search protein 3D structures by collecting structural information from a series of sources, including calculations on the 3D protein structure, sequence annotations in UniproktB and predictions from DAS servers. HOPE works online where one can submit a sequence and mutation only for those that predicted to be damaging by both SIFT and Polyphen (Double Positive) servers. Protein sequences were submitted to project hope server in order to analyze the structural and conformational variations that have resulted from single amino acid substitution corresponding to single nucleotide substitution. Project Hope is available at: http://www.cmbi.ru.nl/hope [17]
GeneMANIA: Helps you to predict the function of your favourite genes and gene sets. GeneMANIA finds other genes that are related to a set of input genes, using a very large set of functional association data. Association data include protein and genetic interactions, pathways, co-expression, co-localization and protein domain similarity. You can use GeneMANIA to find new members of a pathway or complex, find additional genes you may have missed in your screen or find new genes with a specific function, such as protein kinases.
Results and Discussion
APOE gene was investigated in dbSNP/NCBI. APOEgene 7 gene models all of them do not contain SNPs except model 2 XM_005258867.1 which containing a total of 280 SNPs; of which, 5 in 5utr, 98 in near 5utr region, 37 in missense and contig reference, 103 in intron 20 in synonymous, 6 in 3utr region And 13 in near 3 UTR region And only in. Only nsSNPs and 3'UTR SNPs were selected for computational analysis.
Predictions by SIFT and Polyphen
Predictions of deleterious nsSNPs was performed by SIFT and Polyphen softwares; out of 280 snSNPs only 10 (3.5%) were predicted to be damaging by both servers. First, we submitted batch nsSNPs (rs SNPs) to SIFT server; then each of the resultant damaging nsSNPs were submitted to Polyphen as sequences in FASTA Format, it traced 9 probably damaging nsSNPs out of which 6 nsSNPs were marked with the most highest deleterious scores (R50C, L115S, R132S, E139V, E150G, R168H), the other 4 nsSNPs were scored as possibly damaging. Results are shown in the Table 1 below.
Analysis of 3'UTR SNPs by Polymirts (v3.0)
The selected 3'UTR SNPs were 51; all submitted online to PolymiRTS server and resulted in no single pathogenic variant that could cause altered miRNA binding to the 3’UTR and hence no obvious consequences on protein truncation could be expected; despite that we could not excluded possible effects on transcription and splicing efficiency on these regions. This seems to agree with the findings of Karl et al. 2001.
Amino acid substitution effects on protein structure
Project Hope revealed the 3D structure for the truncated proteins with its new candidates; in addition, it described the reaction and physiochemical properties of these candidates. Here we present the results upon each candidate and discus the conformational variations and interactions with the neighbouring amino acids:
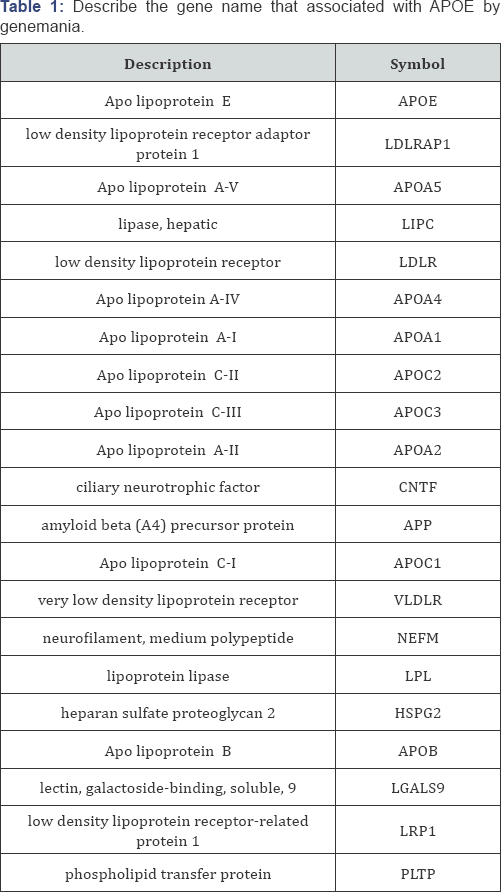
G/A mutation: (rs371694216) resulted in mutation of an Argentine into Histamine at position 43 (R43H). The mutant residue is smaller than the wild-type residue. The wild-type residue was positively charged, the mutant residue is neutral. The wild type residue forms a salt bridge with the Glutamic acid on position 88. The wild type residue forms a salt bridge with the Glutamic acid on position 95. The difference in charge will disturb the ionic interaction made by the original, wild-type residue.
The wild-type residue is not conserved at this position. Another residue type was observed more often at this position in other homologous sequences. This means that other homologous proteins exist with that other residue type than with the wild-type residue in your protein sequence. The other residue type is not similar to your mutant residue. Therefore, the mutation is possibly damaging. Your mutant residue is located near a highly conserved position. There is a difference in charge between the wild-type and mutant amino acid. The charge of the wild-type residue is lost by this mutation. This can cause loss of interactions with other molecules. The wild-type and mutant amino acids differ in size. The mutant residue is smaller than the wild-type residue. This will cause a possible loss of external interactions (Figure 1).
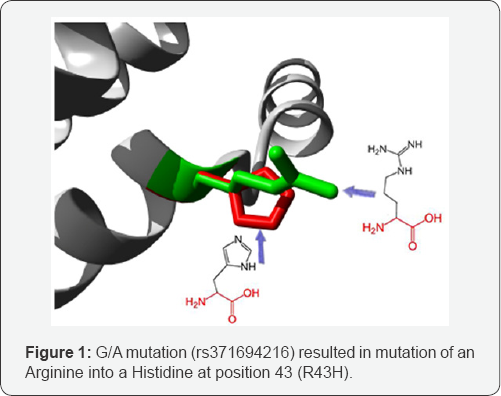
C/T mutation: (rs11542029) Mutation of an Arginine into a Cysteine at position 50. The mutant residue is smaller than the wild-type residue. The wild-type residue was positively charged, the mutant residue is neutral. The mutant residue is more hydrophobic than the wild-type residue. The mutated residue is located on the surface of a domain with unknown function. The residue was not found to be in contact with other domains of which the function is known within the used structure. However, contact with other molecules or domains are still possible and might be affected by this mutation. There is a difference in charge between the wild-type and mutant amino acid. The charge of the wild-type residue is lost by this mutation. This can cause loss of interactions with other molecules. The wild-type and mutant amino acids differ in size. The mutant residue is smaller than the wild-type residue. This will cause a possible loss of external interactions. The hydrophobicity of the wild-type and mutant residue differs (Figure 2).
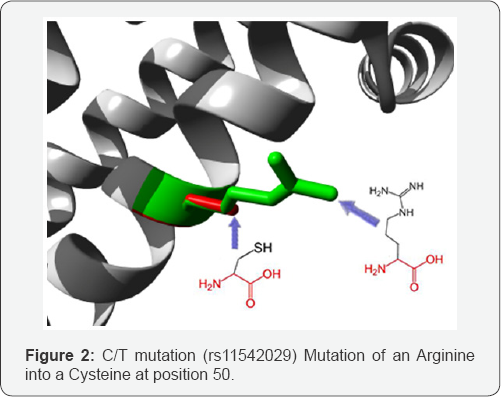
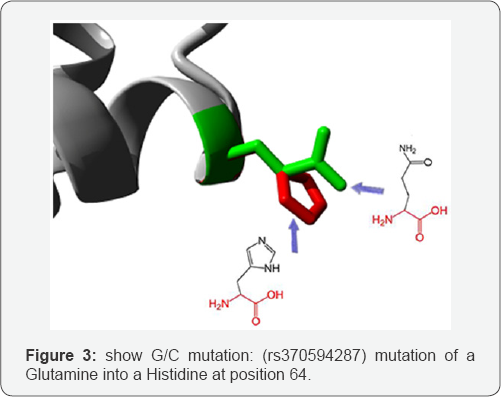
G/C mutation: (rs370594287) mutation of a Glutamine into a Histidine at position 64. The mutant residue is bigger than the wild-type residue. The wild-type residue occurs often at this position in the sequence, but other residues have also been observed here. Your mutant residue was not among the other residue types observed at this position in other, homologous sequences. However, residues with similar properties have been observed, so it is possible that your mutant residue is accepted here too.
The mutated residue is located on the surface of a domain with unknown function. The residue was not found to be in contact with other domains of which the function is known within the used structure. However, contact with other molecules or domains are still possible and might be affected by this mutation. The wild-type and mutant amino acids differ in size. The mutant residue is bigger than the wild-type residue. The residue is located on the surface of the protein; mutation of this residue can disturb interactions with other molecules or other parts of the protein (Figure 3).
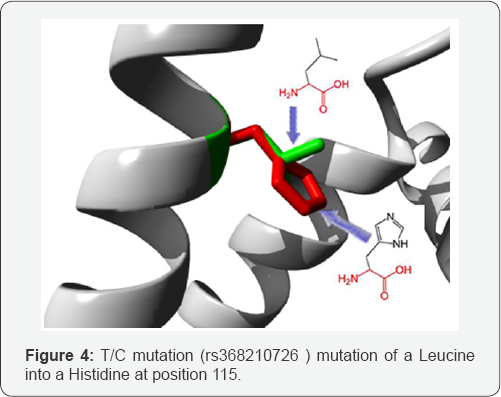
T/C mutation: (rs368210726) mutation of a Leucine into a Histidine at position 115. The mutant residue is bigger than the wild-type residue. The wild-type residue is more hydrophobic than the mutant residue. This residue is part of an interpret domain named 'Apo lipoprotein A/E" (IPR000074). This domain is annotated with the following Gene-Ontology (GO) terms to indicate its function: lipid binding (GO:0008289). More broadly speaking, these GO annotations indicate the domain has a function in binding (GO: 0005488). The mutated residue is buried in a domain that is important for binding of other molecules. The differences between the wild-type and mutant residue might disturb the core structure of this domain and thereby affect the binding properties.
The wild-type and mutant amino acids differ in size. The mutant residue is bigger than the wild-type residue. The wild- type residue was buried in the core of the protein. The mutant residue is bigger and probably will not fit. The hydrophobicity of the wild-type and mutant residue differs. The mutation will cause loss of hydrophobic interactions in the core of the protein (Figure 4).
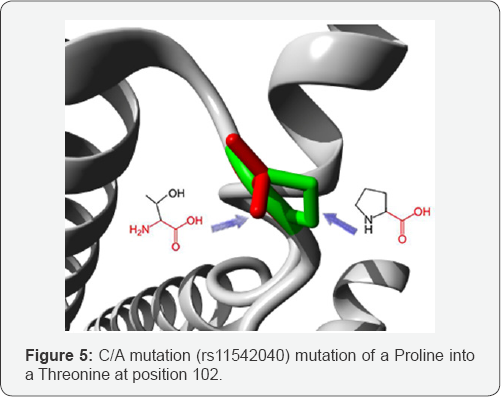
C/A mutation: (rs11542040) mutation of a Proline into a Threonine at position 102. The wild-type residue is more hydrophobic than the mutant residue. A mutation to "Arginine" was found at this position. This variant differs from your mutation but can still be interesting. See the ExPASy site about this variant: VAR_000650. The hydrophobicity of the wild-type and mutant residue differs. The mutation might cause loss of hydrophobic interactions with other molecules on the surface of the protein (Figure 5).
C/A mutation: (rs11542041) mutation of an Arginine into a Serine at position 132. The mutant residue is smaller than the wild-type residue. The wild-type residue was positively charged, the mutant residue is neutral. The mutant residue is more hydrophobic than the wild-type residue. The mutation is located within a stretch of residues annotated in Uniprot as a special region: "8 X 22 AA approximate tandem repeats". The differences in amino acid properties can disturb this region and disturb its function. The mutation is located within a stretch of residues that is repeated in the protein, this repeat is named "3". The mutation into another residue might disturb this repeat and consequently any function this repeat might have.
There is a difference in charge between the wild-type and mutant amino acid. The charge of the wild-type residue is lost by this mutation. This can cause loss of interactions with other molecules. The wild-type and mutant amino acids differ in size. The mutant residue is smaller than the wild-type residue. This will cause a possible loss of external interactions. The hydrophobicity of the wild-type and mutant residue differs (Figure 6).

A/T mutation: (rs41382345) mutation of a Glutamic acid into a Valine at position 139. The mutant residue is smaller than the wild-type residue. The wild-type residue was negatively charged, the mutant residue is neutral. The mutant residue is more hydrophobic than the wild-type residue. The mutation is located within a stretch of residues annotated in Uniprot as a special region: "8 X 22 AA approximate tandem repeats". The differences in amino acid properties can disturb this region and disturb its function. The mutation is located within a stretch of residues that is repeated in the protein, this repeat is named "3". The mutation into another residue might disturb this repeat and consequently any function this repeat might have.
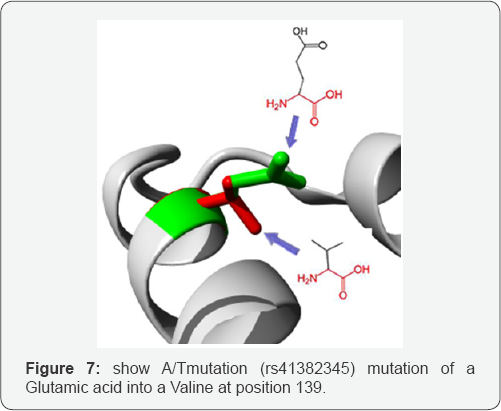
There is a difference in charge between the wild-type and mutant amino acid. The charge of the wild-type residue is lost by this mutation. This can cause loss of interactions with other molecules. The wild-type and mutant amino acids differ in size. The mutant residue is smaller than the wild-type residue. This will cause a possible loss of external interactions. The hydrophobicity of the wild-type and mutant residue differs (Figure 7).
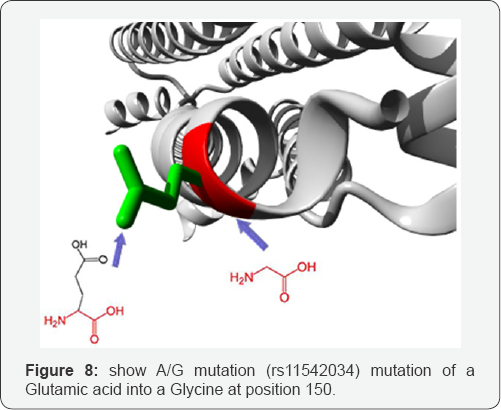
A/G mutation: (rs11542034) mutation of a Glutamic acid into a Glycine at position 150. The mutant residue is smaller than the wild-type residue. The wild-type residue was negatively charged, the mutant residue is neutral. There is a difference in charge between the wild-type and mutant amino acid. The charge of the wild-type residue is lost by this mutation. This can cause loss of interactions with other molecules. The wild- type and mutant amino acids differ in size. The mutant residue is smaller than the wild-type residue. This will cause a possible loss of external interactions. The hydrophobicity of the wild- type and mutant residue differs (Figure 8).
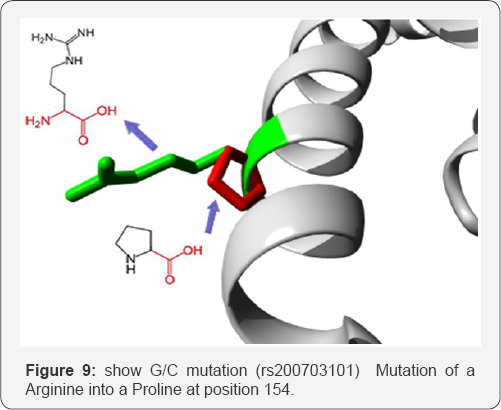
G/C mutation: (rs200703101) Mutation of an Arginine into a Proline at position 154. The mutant residue is smaller than the wild-type residue. The wild-type residue was positively charged, the mutant residue is neutral. The mutant residue is more hydrophobic than the wild-type residue. The wildtype residue forms a salt bridge with the Glutamic acid on position 139. The wild type residue forms a salt bridge with the Glutamic acid on position 150. The difference in charge will disturb the ionic interaction made by the original, wild-type residue. The mutation is located within a stretch of residues annotated in Uniprot as a special region: "8 X 22 AA approximate tandem repeats". The differences in amino acid properties can disturb this region and disturb its function. The mutation is located within a stretch of residues that is repeated in the protein, this repeat is named "4". The mutation into another residue might disturb this repeat and consequently any function this repeat might have. In the 3D-structure can be seen that the wild-type residue is located in a-helix. Proline disrupts a-helix when not located at one of the first 3 positions of that helix. In case of the mutation at hand, the helix will be disturbed and this can have severe effects on the structure of the protein.
Variants
A mutation to "Cysteine" was found at this position. This variant differs from your mutation but can still be interesting. See the ExPASy site about this variant: VAR_000657. A mutation to "Serine" was found at this position. This variant differs from your mutation but can still be interesting. See the ExPASy site about this variant: VAR_000656.
Conservation
The wild-type residue is much conserved, but a few other residue types have been observed at this position too. Neither your mutant residue nor another residue type with similar properties was observed at this position in other homologous sequences. Based on conservation scores this mutation is probably damaging to the protein. There is a difference in charge between the wild-type and mutant amino acid. The charge of the wild-type residue is lost by this mutation. This can cause loss of interactions with other molecules. The wild-type and mutant amino acids differ in size. The mutant residue is smaller than the wild-type residue. This will cause a possible loss of external interactions. The hydrophobicity of the wild-type and mutant residue differs (Figure 9).
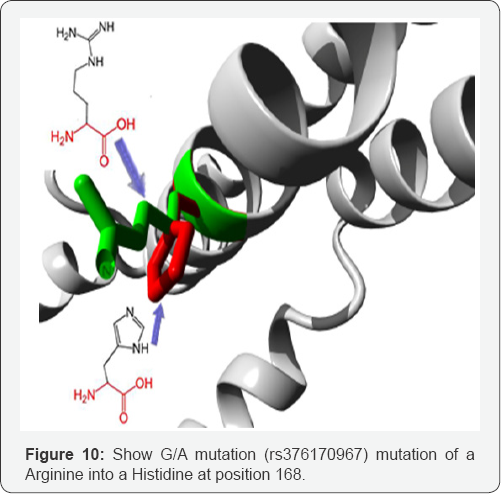
G/A mutation
(rs376170967) mutation of an Arginine into a Histidine at position 168. The mutant residue is smaller than the wild- type residue. The wild-type residue was positively charged, the mutant residue is neutral. There is a difference in charge between the wild-type and mutant amino acid. The charge of the wild-type residue is lost by this mutation. This can cause loss of interactions with other molecules. The wild-type and mutant amino acids differ in size. The mutant residue is smaller than the wild-type residue. This will cause a possible loss of external interactions. Your mutant residue is located near a highly conserved position (Figure 10). In Figure 11 we show the interactions between the gene APOE gene with other gene which mainly function at the lipid metabolism area and in Table 2 the pathway gene names which done by genemania.


Conclusion
The APOE gene was investigated in this work by evaluating the influence of functional SNPs through computation methods, out of a total of 280 SNPs in the APOE, 204 were found to be non- synonymous and 229 were found to be in the 3' untranslated regions, 10 nsSNPs were found to be deleterious and damaging by both Polyphen and SIFT server, 6 SNPs in the 3' UTR were found to be of functional significance. In order to make effective use of genetic diagnosis, the harm SNPs in all Alzheimer's genes should be well known and available to the diagnostic services and molecular biology laboratories to ensure accurate diagnosis for this complicated disease which can also lead to successful intervention which dependent on finding the cause or causes of a problem. From these results we can conclude the APOE gene is very important in the diagnosis of mutation caused by Alzheimer’s disease.
Acknowledgment
The authors wish to acknowledge the enthusiastic cooperation of Africa City of Technology, Sudan.
References
- Shaheen E Lakhan (2017) Alzheimer Disease. Medscape.
- Kasper D, Harrison T (2005) Harrison's principles of internal medicine. New York: McGraw-Hill, Medical Pub. Division.
- http://www.snpedia.com/index.php/Rs429358.
- Serrano-Pozo A, Frosch M, Masliah E, Hyman B (2001) Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb Perspect Med 1(1): a006189-a006189.
- Tanzi R, Bertram L (2005) Twenty Years of the Alzheimer's disease Amyloid Hypothesis: A Genetic Perspective. Cell 120(4): 545-555.
- http://www.abcam.com/human-beta-amyloid-1-42-full-length- protein-ab82795.html
- Saunders A, Strittmatter W, Schmechel D, St. George-Hyslop P, Pericak- Vance M, et al. (1993) Association of apolipoprotein E allele 4 with late-onset familial and sporadic Alzheimer's disease. Neurology 43(8): 1467-1467.
- Agosta F, Vossel K, Miller B, Migliaccio R, Bonasera S, et al. (2009)Apolipoprotein E 4 is associated with disease-specific effects on brain atrophy in Alzheimer's disease and frontotemporal dementia. Nat Rev Neurol 106(6): 2018-2022.
- http://diseasome.kobic.re.kr/pageController.jsp?gene_id=348
- Van der Flier W, Schoonenboom S, Pijnenburg Y, Fox N, Scheltens P (2006) The effect of APOE genotype on clinical phenotype in Alzheimer disease. Neurology 67(3): 526-527.
- Prince J, Zetterberg H, Andreasen N, Marcusson J, Blennow K (2004) APOE 4 allele is associated with reduced cerebrospinal fluid levels of A 42. Neurolog 62(11): 2116-2118.
- http://www.abcam.com/Apolipoprotein-E-antibody-ab20874.html
- http://www.ncbi.nlm.nih.gov/snp/
- http://www.uniprot.org
- http://sift.jcvi.org
- http://genetics.bwh.harvard.edu/pph2/index.shtml
- http://www.cmbi.ru.nl/hope.















