




Modeling of Gene Regulatory Networks Using State Space Models
Samarendra Das*
Department Council of Agricultural Research, Agricultural Statistics Research Institute, India
Submission: March 30, 2017; Published: May 30, 2017
*Corresponding author: Samarendra Das, Department Council of Agricultural Research, Agricultural Statistics Research Institute, India, Email: Samarendra.das@icar.gov.in
How to cite this article: Samarendra D. Modeling of Gene Regulatory Networks Using State Space Models. Curr Trends Biomedical Eng & Biosci. 2017; 4(5): 555646. DOI: 10.19080/CTBEB.2017.04.555646
Abstract
Modeling of gene regulatory networks is becoming popular to understand the complex molecular mechanism of gene regulation due to the availability of high throughput genomic data. Such techniques have encouraged the researchers to understand not only the structure of gene regulatory networks and proteomicnetworks but also gene-gene associations or interactions. State space models are a relatively new approach to infer gene regulatory networks. It has the unique feature of capturing the dynamicity of the gene regulation which is inherent to the biological networks as well as computationally efficiency. The nonlinear state space models also considered the non-linear associations between genes, which all linear modeling approaches fails to capture. Performance evaluation criteria for the approaches used for modeling genetic regulatory networks are also discussed.
Keywords: Gene regulatory networks; State space models; Gene expression value; Micro-array; dynamic modeling
Introduction
Computational Genomics is now becoming the growing area for researchers to decipher biology from genome sequences and related high throughput data. In the post genomic era, there is huge amount of genomic data available because of different advanced experimental technology like microarray technology, Chromatin immune-precipitation with array hybridization (ChIP-chip) etc. [1]. In order to analyze and getting informative knowledge from these data, the efficient statistical approaches are required. These methods are very useful in knowing the interactions among different genes through their by-products, which ultimately regulate the expression of thousand genes in a living cell. Of all the available datasets, gene expression data is the most widely used for gene regulatory network inference. Regulation of gene expression is primarily mediated by regulatory proteins called Transcription Factors (TF). These DNA binding proteins bind to the promoter regions at the start of the genes and thereby controlling the transcription of the genes which ultimately regulates the expression of that gene [2] More specifically, TFs regulate the initiation of transcription through different strategies operating on the transcription mechanism. The interactions between the genes give rise to a complex network like structure known as Gene RegulatoryNetworks(GRN). The understanding of the nature of information on genes and their regulators is improved by the use of network theory, which permits us to uncover some patterns present in gene regulation [3]. Formally, GRNs are modelled as directed graphs which are composed of vertices or nodes as bio-molecules (e.g. genes) and directed edges (connection between genes) represent the regulatory interaction (activatory or inhibitory type).
There are two types of gene expression experiments. In static gene expression experiments, a snapshot of the expression of genes in different samples is measured. While in time series expression experiments, cellular process is measured over time, which leads to generation of time-series gene expression data. The static approaches assume that the genes are expressed in a steady state and thus cannot exploit and describe the dynamic process of gene regulatory process [4] because of changes in mRNA concentration over time during the cell cycle. Rather, the structure of genetic regulatory network is highly dynamic, in the sense that edges may present or absent in the response to different stress conditions over time [5]. Hence, the dynamic modelling GRN is a valuable tool to understand the complexity of gene regulation.There are two types of gene expression experiments. In static gene expression experiments, a snapshot of the expression of genes in different samples is measured. While in time series expression experiments, cellular process is measured over time, which leads to generation of time-series gene expression data. The static approaches assume that the genes are expressed in a steady state and thus cannot exploit and describe the dynamic process of gene regulatory process [4] because of changes in mRNA concentration over time during the cell cycle. Rather, the structure of genetic regulatory network is highly dynamic, in the sense that edges may present or absent in the response to different stress conditions over time [5]. Hence, the dynamic modelling GRN is a valuable tool to understand the complexity of gene regulation.
Types of Biological Data
The advent of advanced experimental technology like microarray technology, Chromatin immune-precipitation with array hybridization (ChIP-chip) leads to the production of huge amount of biological datasets which are quite heterogeneous in nature and difficult to analyse [6]. It can be envisaged that these data sets are very useful in getting insight into the complex underlying mechanism of gene-gene interactions, when they are coupled with suitable statistical techniques. This section reviews some of the main types of data used for the inference of gene regulatory networks.
Micro-array data
Micro-array data are mostly generated by DNA micro-array technology. Microarrays may be used to measure gene expression in many ways, but one of the most popular applications is to compare expression of a set of genes from a cell maintained in a stress or disease condition (condition A) to the same set of genes from a reference cell maintained under normal conditions (condition B).The number of data samples is in general much smaller than the number of genes. The microarray observations are commonly perceived as being extremely noisy [7] because of sources like systematic noise and random noise. The random noise is related to uncontrollable factors (e.g. array effect, biological sample effect etc.) in the microarray experiment and systematic noise (e.g. dye bias and faulty machine readings etc.) may be removed by normalisation.
Gene expression data
Gene expression data is the most widely used for gene regulatory network inference. The level of gene expression is an important indicator of how active a gene is, and is measured in the form of gene expression data. Similarity in the gene expression profiles of two genes advocates some level of correlation between them. So, it is a challenge to remove the error component from the micro-array observations, to get the true gene expression levels of the genes.
Dynamic modelling and inferring gene regulatory networks
Gene regulatory networks exhibit the regulatory relationships (activator or inhibitory) present among the genes. The true construction and interpretations of GRN depend on accurate and reliable estimation of gene-gene associations, which has paramount applications in Computational Biology in general and Medicinal Biology in particular. The following sectionis devoted to the main statistical methods used for dynamic modelling and inference of gene regulatory networks.Gene regulatory networks exhibit the regulatory relationships (activator or inhibitory) present among the genes. The true construction and interpretations of GRN depend on accurate and reliable estimation of gene-gene associations, which has paramount applications in Computational Biology in general and Medicinal Biology in particular. The following sectionis devoted to the main statistical methods used for dynamic modelling and inference of gene regulatory networks.
Linear state space models
The simplest state-space model representation of the genetic regulatory network is given by Wu et al. [8].
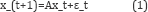
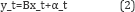
where Ais a matrix representing the regulatory interactions between the genes. y_t and x_tis the observed micro-array and gene expression value of the gene at time t respectively. The noise components ε_tand α_trepresent the transition system and the measurement noise, respectively, and are assumed to be Gaussian. For ease of calculation, matrix Bin equation 2 is usually taken as the identity matrix. Inference as well as estimation of parameters of GRN in case above state-space representation can be performed by expectation-maximization (EM) algorithm [8] and Kalman filter updates [7,9]. The simplicity of the state-space model avoids over-fitting of the network and therefore provides reliable results.
Non-linear state space models
It may be useful to represent genetic networks by simple state-space models, to ease out computational complexity, but the main drawback of the approach is that it almost silent about the non-linear interaction component, which is inherent in biological systems [10]. The state-space model for a time-series consists of a measurement equation relating the observed data to a state vector and a transition equation that describes the evolution of the state vector over time.


where g(.) is a function describing the evolution of the states and h (.) is the function mapping the unknown state vector to the observations. It is assumed that the noise presents in the system 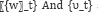 are Gaussian with zero mean and variance- covariance structure is given by
are Gaussian with zero mean and variance- covariance structure is given by
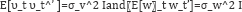
The nonlinear state-space representation model capturing the gene-gene interactions is given by Noor et al. [7]:
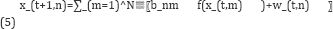
y_t=x_t+u_t (6)
Where N is the total number of genes in the network and f (.) is the regulatory function. The constants b_nmin the model (5) quantify the activation or repression of the expression of the n-th gene by its m-th regulators in the TRN A particular function that is frequently used to capture the nonlinear gene-gene interactions, is the sigmoid squash function given in equation 7 [7,11].
f(x_(t,m) )=1/(1+eA(-x_(t,m) ) ) (7)
One way of solving these equations is by using the Extended Kalman Filter (EKF) [12] which is a popular algorithm for solving nonlinear state-space equations. EKF algorithm provides the solution by approximating the nonlinear system by its first- order linear approximation. Other variants a Kalman filter algorithm like the Cubature Kalman Filter (CKF), Unscented Kalman Filter (UKF) [7].
Performance evaluation
The performance of these approaches can be accessed or evaluated using the knowledge of true network or gold standard network. Given two nodes in a network and the edge prediction problem can have four possible outcomes when compared with the true network (gold standard network): (a) if the edge occurs in both the true and the predicted network, the prediction is called a True Positive (TP), (b) if the edge is predicted but does not occur in the true network, it is a false positive (FP), (c) if the edge does neither occur in the true network nor in the predicted one, it is a True Negative (TN), (d) if the edge occurs in the true network, but is not predicted, it is a False Negative (FN). Once the TP, TN, FN, FP events are counted, it is possible to calculate The True Positive Rate (TPR) and False Positive Rate (FPR)

The accuracy of the approaches can be accessed using the knowledge of true networking. Two indices Area Under the Precision-Recall curve (AUPR) (Plot s Precision vs. Recall) and Area Under Receiver Operator Characteristic Curve (AUROC) (ROC plots FPR vs. TPR) can be used for this purpose.
Conclusion
Several statistical methods have been proposed to model and infer an integrated network of gene regulation. A state-space model for network inference involves parameter estimation which indicates the strength of the activatory and inhibitory regulations among genes. The main feature of state space models is that it captures dynamic structure of gene regulatory mechanisms. The computational efficiency of the linear state space models for the inference of GRN is much higher than that of Dynamic Bayesian technique for network inference [8]. Linear state-space model has the drawback, that it does not throw any light on the non-linear interactions among gene, which can be achieved by adopting nonlinear state space models. Moreover, microarray expression profiles are considered to be highly(9) noisy due to different sources of variation exist in microarray experiments [7]. For this, the measurement equation of state space representation provides the precise estimate gene expression values, which can be further used[13-15].
References
- Panse C, Kshirsagar M (2013) Survey on modelling methods applicable to gene regulatory networks. Int Jour on Bioinf and Biostat 3(3):13-21
- Zhang X, Cheng W, Listgarten J, Kadie C, Huang S (2012) Learning transcriptional regulatory relationships using Sparse Graphical Models. PLoS ONE 7(5): e35762.
- Babu MM, Luscombe NM, Arvind L, Gerstein M, Teichmann SA, at al, (2004) Structure and evolution of transcriptional regulatory networks Curr Opin Struct Biol 14: 283-291
- Tang, B, Hsu HK, Hsu PY, Bonneville R, Chen SS, et al, (2012) Hierarchical modularity in ERa transcriptional network is associated with distinct functions and implicates clinical outcomes Sci Rep 2: 875-897
- Ho JWK, Charleston MA (2011) Network modelling of gene regulation. Biophys Rev 3(1): 1-13.
- Zhou X, Wang E R, Dougherty (2006) Genomic Networks: Statistical Inference from Microarray Data, John Wiley.
- Noor A, Serpedin E, Nounou M, Nounou HN (2012) Inferring gene regulatory networks via nonlinear state-space models IEEE/ACM Trans. Comp Biol Bioinform 9(4): 1203-1211
- Wu X, Li P, Wang N, Gong P, Perkins EJ, et al. (2011) State Space Model with hidden variables for reconstruction of gene regulatory networks BMC Systems Biology 5(Suppl 3): S3
- Xiong J, Zhou T (2013) A Kalman-Filter based approach to identification of time-varying gene regulatory networks. PLoS ONE 8(10): e74571.
- Quach MN, Brunel N, D'alche-Buc F (2007) Estimating parameters and hidden variables in non-linear state-space models based on ODEs for biological networks inference. Bioinformatics 23(23): 3209-3216
- Chen BS, Yang SK, Lan CY, Chuang YJ (2008) A systems biology approach to construct the gene regulatory network of systemic inflammation via microarray and databases mining. BMC Medical Genomics 1: 46
- Wang Z, Liu X, Liu Y, Liang J, Vinciotti V (2009) An extended Kalman filtering approach to modeling nonlinear dynamic gene regulatory networks via short gene expression time series IEEE/ACM Transactions on Computational Biology and Bioinformatics 6(3): 410-419.
- De Jong H (2002) Modeling and simulation of genetic regulatory systems: A Literature Review J Comput Biol 9: 69-103
- Hiros S, Yoshida R, Imoto S, Yamaguchi R, Higuchi T, et al. (2008) Statistical inference of transcriptional module-based gene networks from time course gene expression profiles by using state space models. Bioinformatics 24(7): 932-942
- Zhang SQ, Ching WK, Tsing NK, Leung HY, Guo D (2010) A new multiple regression approach for the construction of genetic regulatory networks Artificial intelligence in medicine 48: 153-160















