



Blood-Drop Liquid Biopsy for Monitoring Mutation Load and Therapeutic Responses
Nader Javadi1, Andrew Ford2 and Chen-Hsiung Yeh2
1Hope Health Center, Reseda, USA
2Circulogene Theranostics, Birmingham, USA
Submission: May 23, 2016; Published: June 10, 2016
*Corresponding author: Chen-Hsiung Yeh, Circulogene Theranostics, 3125 Independence Dr., Suite 301, Homewood, AL 35209 USA, Tel: 1-205-234-0128; Email:cyeh@circulogene.com
How to cite this article: Nader J, Andrew F, Chen-Hsiung Y. Blood-Drop Liquid Biopsy for Monitoring Mutation Load and Therapeutic Responses. Canc Therapy & Oncol Int J. 2016; 1(5): 555572. DOI: 10.19080/CTOIJ.2016.01.555572
Abstract
Tumor heterogeneity, especially intra-tumor heterogeneity, is a crucial factor underlying difficulties in cancer treatment and failure of a number of current therapeutic modalities, even of molecularly targeted therapies. The conventional follow-up care is based on regular observation of protein markers in combination with computed tomography (CT)/positron emission tomography (PET) imaging to monitor stable disease or tumor progression. More recently, the implementation of a noninvasive “liquid biopsy” has been destined to capture tumor-specific genetic clonal evolution throughout the course of cancer therapy. This case report focuses on circulating cell-free tumor DNA (ctDNA) in the bloodstream as a versatile biomarker, enabling accurate monitoring of tumor burden and treatment response. Our observations showed that [1] mutation load measured by blood-drop ctDNA sequencing correlated well with PET/CT, CA biomarkers and clinical outcome in response to therapy; [2] blood-drop ctDNA testing could detect new tumor mass earlier than PET/CT; [3] blood-drop liquid biopsy possessed longitudinal and real-time monitoring capability; [4] blood-drop liquid biopsy could be a surrogate for accurate, affordable and accessible personalized testing.
Keywords: Blood-Drop; Liquid Biopsy; Cell-free tumor DNA; Cancer Therapy
Abbreviations: CT: Computed Tomography; PET: Positron Emission Tomography; ctDNA: Cell-Free Tumor DNA
Introduction
Tumor genome sequencing to inform treatment decisions is the current standard-of-care to personalized cancer management [1,2]. Tumor tissue biopsy, generally from the primary site, is used to determine molecular alterations at a single time point on a single site, before targeted treatment commences. These biopsies carry potential risks for patients, they are painful, costly with high failure rate and unpredictable turnaround time. Most importantly, given the complexities of tumor dynamic heterogeneity, both within a tumor and between a primary tumor and metastases, a tissue sample may not be a true representation of tumor genomic evolution [3-5].
Therefore, inability to capture spatial and temporal heterogeneity during tumor evolution results in the failure of cancer systemic treatments, requiring the development of novel approaches to better track tumor heterogeneity.
Circulating protein biomarkers are traditionally used in cancer diagnosis and in the assessment of therapeutic responses,such as carcinoma embryonic antigen (CEA), prostate-specific antigen (PSA), cancer antigen (CA) 3-15, 9-19, 27-29, and CA-125. Unfortunately, the specificity and reliability of these markers are not satisfactory, and many malignancies even do not have released any reliable protein biomarker [6,7].
The advantage of clinical PET/CT imaging for monitoring tumor response replies upon its ability to interrogate both anatomic and functional measures of treatment effects. Although the resolution of this technology has been greatly improved over years, the reality is even a tiny lesion (approx. 1 cm3) spotted on the image could represent billions of tumor cells.
Circulating cell-free tumor DNA (ctDNA) carries comprehensive, inherently specific, and highly sensitive information, and thus possesses distinctive advantage over conventional protein biomarkers and imaging technology. Studies in melanoma [8], breast [9], ovarian [10], and colon cancers [11] have validated the potential applications of ctDNA to monitor tumor burden dynamically and precisely during treatment process. A liquid biopsy based on ctDNA could capture the entire heterogeneity of the disease longitudinally as well as in real time [12].
This will allow clinicians to ensure that the therapy they have selected, based on a particular molecular target, remains relevant and observe the emergence of any resistance [13]. Instead of waiting for information from imaging scans “reactively”, doctors could “proactively” identify at an earlier stage whether a treatment is not working and to spare the patient with the unnecessary toxicity of a drug that no longer provides any benefit. Likewise, physicians could monitor if any new molecular targets emerging and adjust treatment strategy accordingly. This global picture not only can help for designing combination treatments to minimize therapeutic resistance, but also help to provide patients with the right treatment for the right target without delay.
Materials and Methods
Serum CEA, CA 19-9, 15-3, 27-29, and CA-125 levels were determined by quantitative immunoassay. Blood-drop cellfree DNA was enriched from 20 uL of plasma using Circulogene proprietary technology. DNA concentrations were measured by Qubit fluorometer using dsDNA BR kit. Optimal concentration of 1-10 ng was used to amplify 207 targeted loci using AmpliSeq Cancer Hotspot Panel, version 2 (Life Technologies, Carlsbad, CA, USA), targeted for 2855 hotspot mutations within the 50 cancer driver genes, according to the supplier’s protocol. Subsequent semiconductor-based sequencing was performed on Ion Chef and Ion Proton (Life Technologies, Carlsbad, CA, USA), maintaining the number of reads as >200,000 per sample. Base calling and data analysis was performed as described [14].
Results and Discussion
We performed longitudinal monitoring of somatic alterations in plasma ctDNA, i.e., mutation load, during therapy in 2 patients. The dynamics of mutation load was plotted and compared with CA markers and PET/CT imaging over the course of treatment. Imaging scans were evaluated using the Response Evaluation Criteria in Solid Tumors (RECIST; [15]).
Details of individual cases are provided below.
Patient #1
A 69-year-old non-smoker Chinese female patient with diagnosis of lung cancer with metastasis. Tissue biopsy showed sensitizing EGFR mutation, while ALK, ROS-1, BRAF were negative on May 19, 2015. She was then placed on Tarceva 100 mg daily and continued with Avastin once a month. After near 5-month of targeted drug administration, her CEA, CA19-9, CA125 levels all dropped significantly (Figure 1, left panel), and PET/CT scan evaluation also showed stable disease that was maintained for 101 days with decreases in tumor size and activity (Figure 1, right panel).
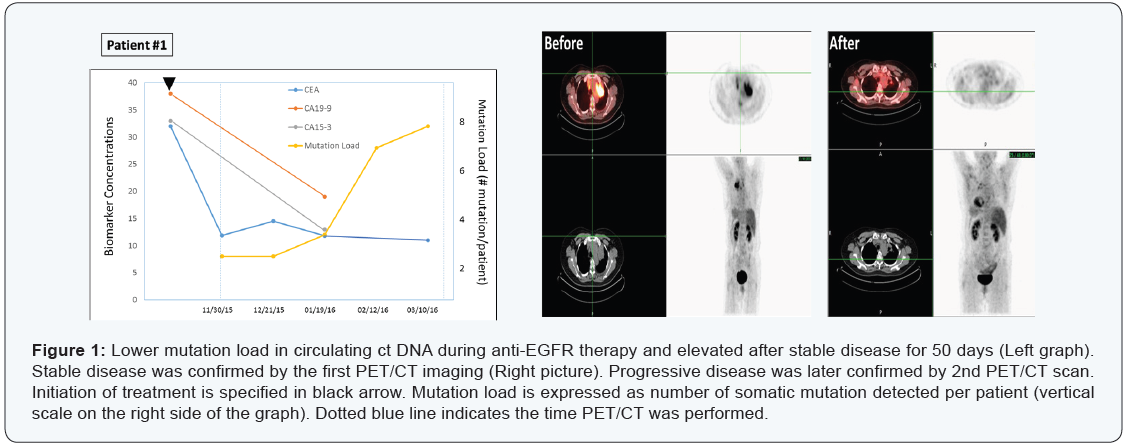
On the other hand, plasma ctDNA mutation dynamics detected a new progression. Two somatic mutations was maintained for about 50 days (in TP53 and PTEN genes), then gradually increased to 3, 7 and 8 mutations by the end of March 2016, indicating a progressive disease.
The mutation analysis on March 10th revealed 2 low-allelefrequency sub clones of EGFR - E114K (4.2%) and E868G (2.4%), implying a clonally evolution upon selection pressure by the drugs. Most importantly, PET/CT scans on March 31, 2016 identified a new tumor mass (about 1 cm3), confirming what ct DNA mutation analysis has found earlier (Figure 1) upper panel.
Overall, this case demonstrated that the ctDNA “genetic responses” were closely associated with radiologically stable disease, with increases in the mutation load emerging ~2 months earlier than radiological progression.
Patient #2
A 79-year-old Iranian female patient with diagnosis of metastatic peri-pancreatic lymph node adenocarcinoma with unknown primary. Immunohistochemistry on fine-needle biopsy revealed CK7 positive, while CK20, TTF, S100 and CD45 all negative on October 26, 2015. She was placed on XELOX initially. Later immunostaining also identified PD-L1 overexpression, therefore, she was then treated with XELIRI, Avastin, and Opdivo. PET/CT scans on February 11, 2016 showed significant decrease in tumor size and activity with >90% response rate (Figure 2, right panel).
Results from mutation load by plasma ctDNA sequencing were in agreement with those of imaging, CEA and CA markers (CA125, CA27-29, CA19-9), indicating a stable disease maintained at least for 41 days (Figure 2, left panel).
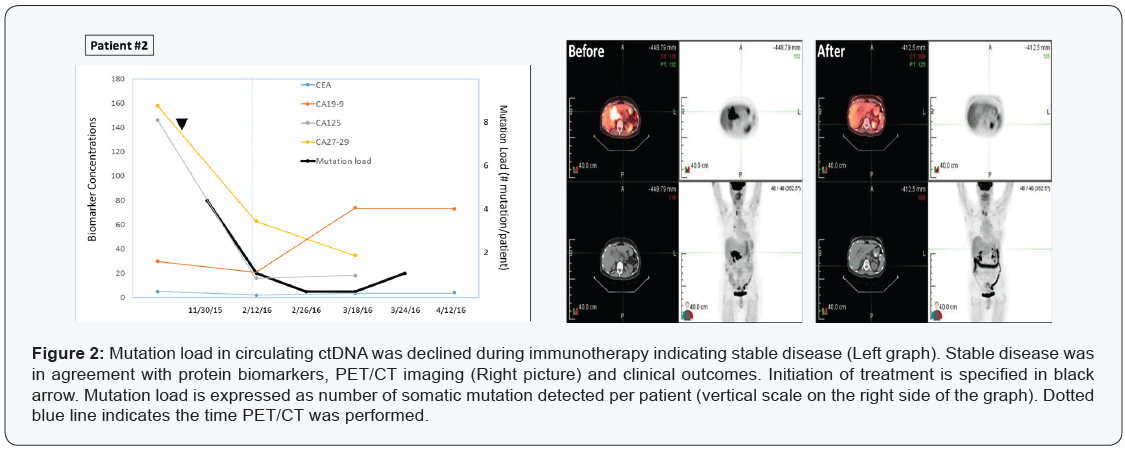
Four somatic mutations were detected initially (FLT3 Y572C 6.5%; TP53 E165G 5.2%; TP53 Y104C 4.1%; TP53 C137Y 4.0%), then declined to 1 and zero mutation during the course of treatment. This case illustrated again the mutation analysis by blood-drop liquid biopsy correlated strongly with clinical outcomes in response to therapy.
The CEA and CA protein markers are not necessarily specific to cancer cells and PET/CT scans are suffering from resolution limitation. By contrast, cancer-associated somatic mutations are specific to malignancies, and plasma DNA with these mutations is indicative for the presence of malignancies. Our results on these two cases support the notion that levels of the somatic mutations detected from blood correlated well with current standard care test results and clinical outcomes, i.e., stable disease or progressive disease, and may provide the earliest indication of recurrence.
Conclusion
We have demonstrated for the first time that blood-drop ctDNA sequencing test can detect the presence of new tumor mass earlier than current standard care imaging methods. The mutation detection of ctDNA in drops of blood is a powerful monitoring tool capable of providing accurate and earlier assessment of tumor behavior, burden and patient responses following treatment.
References
- Meric-Bernstam F, Mills GB (2012) Overcoming implementation challenges of personalized cancer therapy. Nat Rev ClinOncol 9(9): 542-548.
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, et al. (2013) Cancer genome landscapes. Science 339(6127):1546-1558.
- Marusyk A, Polyak K (2010) Tumor heterogeneity: causes and consequences. Biochim Biophys Acta 1805(1): 105-117.
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, et al.(2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366(10): 883-892.
- Aparicio S, Caldas C (2013) The implications of clonal genome evolution for cancer medicine. N Engl J Med 368(9): 842-851.
- Yoshimasu T, Maebeya S, Suzuma T, Bessho T, Tanino H, et al. (1999) Disappearance curves for tumor markers after resection of intrathoracic malignancies. Int J Biol Markers 14(2): 99-105.
- Ito K, Hibi K, Ando H, Hidemura K, Yamazaki T, et al. (2002) Usefulness of analytical CEA doubling time and half-life time for overlooked synchronous metastases in colorectal carcinoma. J Clin On col 32(2): 54-58.
- Bidard FC, Madic J, Mariani P, Piperno-Neumann S, Rampanou A, et al. (2014) Detection rate and prognostic value of circulating tumor cells and circulating tumor DNA in metastatic uveal melanoma. Int J Cancer 134(5): 1207-1213.
- Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, et al. (2013) Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 368(13): 1199-1209.
- Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, et al. (2012) Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Trams Med 4(136): 136- 168.
- Tie J, Kinde I, Wang Y, Wong HL, Roebert J, et al. (2015) Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann On col 26(8): 1715-1722.
- Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, et al. (2008) Circulating mutant DNA to assess tumor dynamics. Nat Med 14(9): 985-990.
- Diaz LA , Bardelli A (2014) Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol 32(6): 579-586.
- Spurgin J, Ford A, Athanasuleas J, Yeh CH (2015) Next-generation targeted sequencing of circulating cell-free DNA from droplet volumes of blood. Intl J Life Sci Res 3(3): 23-29.
- Eisenhauera EA, Therasseb P, Bogaertsc J, Schwartzd LH, Sargente D, et al. (2009) New response evaluation criteria in solid tumours. Eur J Cancer 45(2): 228-247.
